
Filed Pursuant to Rule 424(b)(5)
Registration No. 333-229118
PROSPECTUS SUPPLEMENT
(To Prospectus Dated February 4, 2019)
Up to $20,000,000
Common Stock
We have entered into an Open Market Sale AgreementSM, or sales agreement, with Jefferies LLC, or Jefferies, dated December 13, 2019, relating to shares of our common stock, $0.0001 par value per share, offered by this prospectus supplement and the accompanying prospectus. In accordance with the terms of the sales agreement, we may offer and sell shares of our common stock having an aggregate offering price of up to $20,000,000 from time to time through Jefferies acting as sales agent, at our discretion.
Our common stock is listed on The Nasdaq Capital Market under the symbol “AVCO.” On December 12, 2019, the closing sale price of our common stock on The Nasdaq Capital Market was $2.32 per share.
Upon our delivery of a placement notice and subject to the terms and conditions of the sales agreement, Jefferies may sell shares of our common stock by methods deemed to be an “at the market offering” as defined in Rule 415(a)(4) promulgated under the Securities Act of 1933, as amended, or the Securities Act.. Jefferies will act as sales agent using its commercially reasonable efforts consistent with its normal trading and sales practices, on mutually agreed terms between Jefferies and us. There is no arrangement for funds to be received in any escrow, trust or similar arrangement.
Jefferies will be entitled to compensation at a fixed commission rate of 3.0% of the gross proceeds of each sale of shares of our common stock. See “Plan of Distribution” beginning on page S-6 for additional information regarding the compensation to be paid to Jefferies. In connection with the sale of our common stock on our behalf, Jefferies will be deemed to be an “underwriter” within the meaning of the Securities Act and the compensation of Jefferies will be deemed to be underwriting commissions or discounts. We have also agreed to provide indemnification and contribution to Jefferies with respect to certain liabilities, including liabilities under the Securities Act.
Investing in our securities involves a high degree of risk. Before making an investment decision, please read the information under the heading “Risk Factors” beginning on page S-3 of this prospectus supplement and in the documents incorporated by reference into this prospectus supplement and the accompanying prospectus.
Neither the Securities and Exchange Commission nor any state securities commission has approved or disapproved of these securities or passed on the adequacy or accuracy of this prospectus supplement or the accompanying prospectus. Any representation to the contrary is a criminal offense.
Jefferies
December 13, 2019
TABLE OF CONTENTS
| PROSPECTUS SUPPLEMENT | |
| Page | |
| About This Prospectus Supplement | S-ii |
| Cautionary Note Regarding Forward-Looking Statements | S-iii |
| Prospectus Supplement Summary | S-1 |
| Risk Factors | S-3 |
| Use of Proceeds | S-4 |
| Dilution | S-5 |
| Plan of Distribution | S-6 |
| Legal Matters | S-7 |
| Experts | S-7 |
| Where You Can Find More Information | S-7 |
| Incorporation by Reference | S-7 |
| ACCOMPANYING PROSPECTUS | |
| Page | |
| About This Prospectus | 1 |
| About the Company | 2 |
| Recent Development | 18 |
| Risk Factors | 21 |
| Cautionary Note Regarding the Forward-Looking Statements | 49 |
| Use of Proceeds | 50 |
| Dividend Policy | 50 |
| Offering and Listing Details | 50 |
| Description of Capital Stock | 51 |
| Description of Warrants | 52 |
| Description of Units | 53 |
| Income Tax Considerations | 53 |
| Plan of Distribution | 54 |
| Where You Can Find More Information | 55 |
| Incorporation by Reference | 55 |
| Material Changes | 56 |
| Legal Matters | 57 |
| Experts | 57 |
S-i
ABOUT THIS PROSPECTUS SUPPLEMENT
This prospectus supplement and the accompanying prospectus relate to the offering of our common stock. Before buying any of the common stock that we are offering, we urge you to carefully read this prospectus supplement and the accompanying prospectus, together with the information incorporated by reference as described under the heading “Incorporation by Reference” in this prospectus supplement and the accompanying prospectus. These documents contain important information that you should consider when making your investment decision.
This document is in two parts. The first part is this prospectus supplement, which describes the specific terms of this offering and also adds to and updates information contained in the accompanying prospectus and the documents incorporated by reference herein or therein. The second part, the accompanying prospectus, provides more general information. Generally, when we refer to this prospectus, we are referring to both parts of this document combined. To the extent there is a conflict between the information contained in this prospectus supplement and the information contained in the accompanying prospectus or any document incorporated by reference herein or therein filed prior to the date of this prospectus supplement, you should rely on the information in this prospectus supplement; provided that if any statement in one of these documents is inconsistent with a statement in another document having a later date—for example, a document incorporated by reference in the accompanying prospectus—the statement in the document having the later date modifies or supersedes the earlier statement.
We further note that the representations, warranties and covenants made by us in any agreement that is filed as an exhibit to any document that is incorporated by reference herein or in the accompanying prospectus were made solely for the benefit of the parties to such agreement, including, in some cases, for the purpose of allocating risk among the parties to such agreement, and should not be deemed to be a representation, warranty or covenant to you. Moreover, such representations, warranties or covenants were accurate only as of the date when made. Accordingly, such representations, warranties and covenants should not be relied on as accurately representing the current state of our affairs.
You should rely only on the information contained in, or incorporated by reference in, this prospectus supplement, the accompanying prospectus and in any free writing prospectus that we may authorize for use in connection with this offering. We have not, and Jefferies has not, authorized anyone to provide you with any information other than that contained or incorporated by reference in this prospectus supplement, in the accompanying prospectus or in any free writing prospectus prepared by or on behalf of us or to which we have referred you. We and Jefferies take no responsibility for, and can provide no assurance as to the reliability of, any other information that others may give you. We are not, and Jefferies is not, making an offer to sell, or soliciting an offer to purchase, the securities offered by this prospectus supplement and the accompanying prospectus in any jurisdiction to or from any person to whom or from whom it is unlawful to make such offer or solicitation of an offer in such jurisdiction. The information contained in this prospectus supplement, the accompanying prospectus, the documents incorporated by reference herein or therein, and in any free writing prospectus prepared by or on behalf of us that we may authorize for use in connection with this offering is accurate only as of the date of those respective documents. Our business, financial condition, results of operations and prospects may have changed since those dates. It is important for you to read and consider all information contained in this prospectus supplement, the accompanying prospectus, the documents incorporated by reference herein and therein, and any free writing prospectus prepared by or on behalf of us that we may authorize for use in connection with this offering, in their entirety, before making an investment decision. You should also read and consider the information in the documents to which we have referred you in the sections entitled “Where You Can Find More Information” and “Incorporation by Reference” in this prospectus supplement and the accompanying prospectus.
We and Jefferies are offering to sell, and seeking offers to buy, shares of our common stock only in jurisdictions where offers and sales are permitted. The distribution of this prospectus supplement and the accompanying prospectus and the offering of the common stock in certain jurisdictions may be restricted by law. Persons outside the United States who come into possession of this prospectus supplement and the accompanying prospectus must inform themselves about, and observe any restrictions relating to, the offering of the common stock and the distribution of this prospectus supplement and the accompanying prospectus outside the United States. This prospectus supplement and the accompanying prospectus do not constitute, and may not be used in connection with, an offer to sell, or a solicitation of an offer to buy, any securities offered by this prospectus supplement and the accompanying prospectus by any person in any jurisdiction in which it is unlawful for such person to make such an offer or solicitation.
Unless otherwise stated, all references in this prospectus supplement and the accompanying prospectus to “we”, “us”, “our”, “Company”, “our company”, “Avalon GloboCare” and “Avalon” refer to Avalon GloboCare Corp., a Delaware corporation, either alone or together with our consolidated subsidiaries as the context requires.
This prospectus supplement and the accompanying prospectus contains references to our trademarks and to trademarks belonging to other entities. Solely for convenience, trademarks and trade names referred to in this prospectus supplement and the accompanying prospectus, including logos, artwork and other visual displays, may appear without the ® or TM symbols, but such references are not intended to indicate, in any way, that their respective owners will not assert, to the fullest extent under applicable law, their rights thereto. We do not intend our use or display of other companies’ trade names or trademarks to imply a relationship with, or endorsement or sponsorship of us by, any other companies.
S-ii
CAUTIONARY NOTE REGARDING FORWARD-LOOKING STATEMENTS
This prospectus supplement and the accompanying prospectus contain, and the documents incorporated by reference herein and therein and any free writing prospectus that we have authorized for use in connection with this offering may contain, forward-looking statements within the meaning of Section 27A of the Securities Act and Section 21E of the Securities Exchange Act of 1934, as amended, or the Exchange Act. All statements other than statements of historical facts contained in this prospectus supplement, the accompanying prospectus and the information we incorporate by reference are forward-looking statements. These statements relate to future events or to our future operating or financial performance and involve known and unknown risks, uncertainties and other factors which may cause our actual results, performance or achievements to be materially different from any future results, performances or achievements expressed or implied by the forward-looking statements. Forward-looking statements may include, but are not limited to statements about:
| ● | our ability to attract and retain management; |
| ● | our ability to raise capital when needed and on acceptable terms and conditions; |
| ● | the intensity of competition; |
| ● | general economic conditions; |
| ● | changes in regulations; |
| ● | whether the market for healthcare services continues to grow, and, if it does, the pace at which it may grow; and |
| ● | our ability to compete against large competitors in a rapidly changing market. |
In some cases, you can identify forward-looking statements by terms such “anticipate,” “believe,” “estimate,” “expect,” “forecast,” “intend,” “may,” “plan,” “project,” “target,” “will” and other words and terms of similar meaning. These statements reflect our current views with respect to future events and are based on assumptions and subject to risks and uncertainties. Given these uncertainties, you should not place undue reliance on these forward-looking statements. We discuss many of these risks in greater detail under the heading “Risk Factors” in this prospectus supplement and in our SEC filings.
You should read this prospectus supplement, the accompanying prospectus, the documents we have filed with the SEC that are incorporated by reference and any free writing prospectus that we have authorized for use in connection with this offering completely and with the understanding that our actual future results may be materially different from what we expect. We qualify all of the forward-looking statements in the foregoing documents by these cautionary statements. You should not place undue reliance on these statements. Forward-looking statements speak only as of the date of the document containing the applicable statement. We do not undertake any obligation to publicly update any forward-looking statements.
S-iii
This summary highlights certain information about us, this offering and selected information contained elsewhere in or incorporated by reference in this prospectus supplement. This summary is not complete and does not contain all of the information that you should consider before deciding whether to invest in our securities. For a more complete understanding of our company and this offering, we encourage you to read and consider carefully the more detailed information in this prospectus supplement and the accompanying prospectus, including the information included under the heading “Risk Factors” in this prospectus supplement beginning on page S-3, the information included under the heading “Risk Factors” in the accompanying prospectus beginning on page 21, the information incorporated by reference in this prospectus supplement and the accompanying prospectus, which is described under “Where You Can Find More Information” and “Incorporation by Reference,” and the information included in any free writing prospectus that we have authorized for use in connection with this offering.
Overview
We are a clinical-stage, leading CellTech bio-developer dedicated to advancing and empowering innovative, transformative immune effector cell therapy and exosome technology. We also provide strategic advisory and outsourcing services to facilitate and enhance our clients’ growth, development, as well as competitiveness in healthcare and CellTech industry markets.
Our subsidiary and joint venture structure contribute to investor flexibility and research and development focus, enabling us to establish our leading role in the fields of immune effector cell therapy (including CAR-T and CAR-NK), exosome-based regenerative therapeutics (our Avalon Clinical-grade Tissue-specific Exosome, or ACTEXTM, platform), as well as “liquid biopsy” diagnostics.
We achieve and foster seamless integration of unique verticals to bridge and accelerate innovative research, bio-process development, clinical programs and product commercialization.
Our upstream innovative research includes:
| ● | Co-development of ACTEXTM with Weill Cornell Medicine; |
| ● | Novel therapeutic and diagnostic targets development utilizing QTY-code protein design technology with Massachusetts Institute of Technology, orMIT; and |
| ● | Co-development of next generation, transposon-based, multi-target CAR-T, CAR-NK and other immune effector cell therapeutic modalities with Arbele Corp. |
Our midstream bio-processing and bio-production facility is located in Nanjing, China with state-of-the-art, automated GMP and QC/QA infrastructure for standardized bio-manufacturing of clinical-grade cellular products involved in our clinical programs in immune effector cell therapy, regenerative therapeutics, as well as bio-banking.
Our downstream medical team and facility consists of top-rated affiliated hospital network and experts specialized in hematology, oncology, cellular immunotherapy, hematopoietic stem/progenitor cell transplant, as well as regenerative therapeutics. Our major clinical programs include:
| ● | AVA-001: We have initiated our first-in-human clinical trial of our CD19 CAR-T candidate, AVA-001, in August 2019 at the Hebei Yanda Lu Daopei Hospital and Beijing Lu Daopei Hospital in China for the indication of relapsed/refractory B-cell acute lymphoblastic leukemia and non-Hodgkin Lymphoma. AVA-001 is characterized by the utilization of 4-1BB (CD137) co-stimulatory signaling pathway, conferring a strong anti-cancer activity during pre-clinical study. It also features a shorter bio-manufacturing time which leads to advantage of prompt treatment to patients with these dreadful hematologic malignancies. We plan to recruit 20 patients (under registered clinical trial NCT03952523) for safety and efficacy studies. |
| ● | AVA-101: Our transposon-based, multi-targeted CAR-T candidate, AVA-101 (co-developed with Arbele Corp.), has entered the pre-clinical process development and validation phase. AVA-101 features non-viral, transposon-engineered CAR-T with multiple anti-cancer targets, as well as possessing molecular safety-switch mechanism to minimize the side effects, such as cytokine release syndrome and neurotoxicity, often associated with conventional CAR-T cellular therapy. We anticipate this next generation of potentially more effective and safer CAR-T candidate will proceed to a first-in-human clinical study in the later part of 2020. |
| ● | AVA-202: We have recently completed the standardized bio-production process of tissue-specific, clinical-grade exosomes, a co-development endeavor with Weill Cornell Medicine with focus on angiogenic exosomes derived from endothelial cells which promote blood vessel formation and wound healing. We are further developing this technology platform into a therapeutic candidate, AVA-202, and plan to initiate international multi-centered clinical studies in unmet medical areas of vascular diseases and wound healing, including treatment of diabetic foot ulcer. |
The commercialization phase of our ACTEXTM-based product development is underway to enter the markets of skin care, scar removal, and hair growth through in-house development and strategic partnership.
Company Information
We were incorporated under the laws of the State of Delaware on July 28, 2014 under the name Global Technologies Corp. On October 18, 2016, we changed our name to Avalon GloboCare Corp. Our principal executive offices are located at 4400 Route 9 South, Suite 3100, Freehold, New Jersey 07728, and our telephone number is (732) 780-4400. Our Internet website is www.avalon-globocare.com. The information on, or that can be accessed through, our website is not part of this prospectus supplement, and you should not rely on any such information in making the decision whether to purchase our securities.
S-1
The Offering
| Common stock offered by us | Shares of our common stock having an aggregate offering price of up to $20,000,000. |
| Common stock to be outstanding after this offering | Up to 8,620,690 shares, assuming sales at a price of $2.32 per share, which was the closing price of our common stock on The Nasdaq Capital Market on December 12, 2019. The actual number of shares issued will vary depending on the sales price under this offering. |
| Plan of Distribution | “At the market offering” that may be made from time to time through our agent, Jefferies LLC. See the section entitled “Plan of Distribution” on page S-6 of this prospectus supplement. |
| Use of Proceeds | We currently intend to use the net proceeds from this offering to fund our core technology platform development and clinical studies in exosome-based liquid biopsy and regenerative therapeutics, next-generation multi-targeted CAR-T immunotherapy, co-development projects with MIT and Weill Cornell, as well as for working capital and other general corporate purposes. See the section entitled “Use of Proceeds” on page S-4 of this prospectus supplement. |
| Risk Factors | This investment involves a high degree of risk. See “Risk Factors” beginning on page S-3 of this prospectus supplement and the other information included in, or incorporated by reference into, this prospectus supplement for a discussion of certain factors you should carefully consider before deciding to invest in shares of our common stock. |
| Nasdaq Capital Market symbol | AVCO |
The number of shares of our common stock to be outstanding after this offering is based on 75,771,056 shares of our common stock issued and outstanding as of September 30, 2019 and excludes:
| ● | 5,070,000 shares of common stock issuable upon exercise of outstanding stock options at a weighted-average exercise price of $1.43 per share as of September 30, 2019; and |
| ● | 1,714,288 shares of common stock issuable upon exercise of outstanding warrants to purchase common stock at a weighted-average exercise price of $3.50 per share as of September 30, 2019. |
S-2
An investment in our securities involves a high degree of risk. Before deciding whether to invest in our securities, you should consider carefully the risks described below and discussed under the sections captioned “Risk Factors” contained in our Annual Report on Form 10-K for the year ended December 31, 2018 and our Quarterly Report on Form 10-Q for the quarter ended September 30, 2019, which are incorporated by reference in this prospectus supplement and the accompanying prospectus, together with other information in this prospectus supplement, the accompanying prospectus, the information and documents incorporated by reference, and in any free writing prospectus that we have authorized for use in connection with this offering. See “Where You Can Find More Information” and “Incorporation by Reference.” If any of these risks actually occurs, our business, financial condition, results of operations or cash flow could be harmed. This could cause the trading price of our common stock to decline, resulting in a loss of all or part of your investment. The risks described below and in the documents referenced above are not the only ones that we face. Additional risks not presently known to us or that we currently deem immaterial may also affect our business.
Risks Related To This Offering
If you purchase shares of common stock in this offering, you will suffer immediate and substantial dilution of your investment.
The price per share of our common stock in this offering may exceed the net tangible book value per share of our common stock outstanding prior to this offering. Therefore, if you purchase shares of our common stock in this offering, you may pay a price per share that substantially exceeds our net tangible book value per share after this offering. To the extent shares are issued under outstanding options or warrants at exercise prices lower than the price of our common stock in this offering, you will incur further dilution. Assuming that an aggregate of 8,620,690 shares of our common stock are sold at a price of $2.32 per share, the last reported sale price of our common stock on The Nasdaq Capital Market on December 12, 2019, for aggregate proceeds to us of $20,000,000, and after deducting commissions and estimated aggregate offering expenses payable by us, you will experience immediate dilution of $2.03 per share, representing the difference between our as adjusted net tangible book value per share as of September 30, 2019 after giving effect to this offering and the assumed offering price. See the section entitled “Dilution” below for a more detailed illustration of the dilution you would incur if you participate in this offering.
You may experience future dilution as a result of future equity offerings.
In order to raise additional capital, we may in the future offer additional shares of our common stock or other securities convertible into or exchangeable for our common stock at prices that may not be the same as the price per share in this offering. We may sell shares or other securities in any other offering at a price per share that is less than the price per share paid by investors in this offering, and investors purchasing shares or other securities in the future could have rights superior to existing stockholders. The price per share at which we sell additional shares of our common stock, or securities convertible or exchangeable into common stock, in future transactions may be higher or lower than the price per share paid by investors in this offering.
An active trading market for our common stock may not be sustained.
Although our common stock is listed on The Nasdaq Capital Market, an active trading market for our shares may not be sustained. If an active market for our common stock does not continue, it may be difficult for you to sell shares you purchase in this offering without depressing the market price for the shares, or at all. An inactive trading market for our common stock may also impair our ability to raise capital to continue to fund our operations by selling shares and may impair our ability to acquire other companies or technologies by using our shares as consideration.
We have broad discretion in the use of our cash and cash equivalents, including the net proceeds we receive in this offering, and may not use them effectively.
Our management will have broad discretion in the application of the net proceeds we receive in this offering, including for any of the purposes described in the section entitled “Use of Proceeds,” and you will not have the opportunity as part of your investment decision to assess whether our management is using the net proceeds appropriately. Because of the number and variability of factors that will determine our use of our net proceeds from this offering, their ultimate use may vary substantially from their currently intended use. The failure by our management to apply these funds effectively could result in financial losses that could have a material adverse effect on our business and cause the price of our common stock to decline. Pending their use, we may invest our net proceeds from this offering in short-term, investment-grade, interest-bearing securities. These investments may not yield a favorable return to our stockholders.
S-3
We currently intend to use the net proceeds from this offering to fund our core technology platform development and clinical studies in exosome-based liquid biopsy and regenerative therapeutics, next-generation multi-targeted CAR-T immunotherapy, co-development projects with MIT and Weill Cornell, as well as for working capital and other general corporate purposes.
The expected use of net proceeds from this offering represents our intentions based upon our current plans and business conditions, which could change in the future as our plans and business conditions evolve. The amount and timing of these expenditures will depend on a number of factors, including the progress of our research and development efforts, the progress of any partnering efforts, technological advances and the competitive environment for our product candidates. Accordingly, you will be relying on the judgment of our management with regard to the use of these net proceeds, and you will not have the opportunity, as part of your investment decision, to assess whether the proceeds are being used appropriately. It is possible that the proceeds will be used in a way that does not yield a favorable, or any, return for us.
Pending application of the net proceeds as described above, we intend to invest the proceeds in investment grade interest bearing instruments, or will hold the proceeds in interest bearing or non-interest bearing bank accounts.
S-4
If you invest in this offering, your ownership interest will be diluted immediately to the extent of the difference between the public offering price per share and the as-adjusted net tangible book value per share of our common stock after giving effect to this offering. We calculate net tangible book value per share by dividing the net tangible book value, which is tangible assets less total liabilities, by the number of outstanding shares of our common stock. Dilution represents the difference between the portion of the amount per share paid by purchasers of shares in this offering and the as adjusted net tangible book value per share of our common stock immediately after giving effect to this offering. Our net tangible book value as of September 30, 2019 was approximately $5.7 million, or $0.08 per share.
After giving effect to the sale of our common stock pursuant to this prospectus supplement and accompanying prospectus in the aggregate amount of $20,000,000 at an assumed offering price of $2.32 per share, the last reported sale price of our common stock on The Nasdaq Capital Market on December 12, 2019, and after deducting commissions and estimated aggregate offering expenses payable by us, our net tangible book value as of September 30, 2019 would have been approximately $24.8 million, or $0.29 per share of common stock. This represents an immediate increase in the net tangible book value of $0.21 per share to our existing stockholders and an immediate dilution in net tangible book value of $2.03 per share to new investors. The following table illustrates this per share dilution:
| Assumed offering price per share | $ | 2.32 | ||||||
| Net tangible book value per share as of September 30, 2019 | $ | 0.08 | ||||||
| Increase per share attributable to new investors | $ | 0.21 | ||||||
| As-adjusted net tangible book value per share as of September 30, 2019 after giving effect to this offering | $ | 0.29 | ||||||
| Dilution per share to new investors purchasing shares in this offering | $ | 2.03 |
The table above assumes for illustrative purposes that an aggregate of 8,620,690 shares of our common stock are sold pursuant to this prospectus supplement and the accompanying prospectus at a price of $2.32 per share, the last reported sale price of our common stock on The Nasdaq Capital Market on December 12, 2019, for aggregate gross proceeds of $20,000,000. The shares sold in this offering, if any, will be sold from time to time at various prices. An increase of $1.00 per share in the price at which the shares are sold from the assumed offering price of $2.32 per share, assuming all of our common stock in the aggregate amount of $20,000,000 is sold at that price, would result in an adjusted net tangible book value per share after the offering of $0.30 per share and would increase the dilution in net tangible book value per share to new investors in this offering to $3.02 per share, after deducting commissions and estimated aggregate offering expenses payable by us. A decrease of $1.00 per share in the price at which the shares are sold from the assumed offering price of $2.32 per share, assuming all of our common stock in the aggregate amount of $20,000,000 is sold at that price, would result in an adjusted net tangible book value per share after the offering of $0.27 per share and would decrease the dilution in net tangible book value per share to new investors in this offering to $1.05 per share, after deducting commissions and estimated aggregate offering expenses payable by us. This information is supplied for illustrative purposes only.
The above discussion and table are based on 75,771,056 shares of our common stock issued and outstanding as of September 30, 2019 and excludes the following:
| ● | 5,070,000 shares of common stock issuable upon exercise of outstanding stock options at a weighted-average exercise price of $1.43 per share as of September 30, 2019; and |
| ● | 1,714,288 shares of common stock issuable upon exercise of outstanding warrants to purchase common stock at a weighted-average exercise price of $3.50 per share as of September 30, 2019. |
S-5
We have entered into a sales agreement with Jefferies, under which we may offer and sell up to $20.0 million of our shares of common stock from time to time through Jefferies acting as agent. Sales of our shares of common stock, if any, under this prospectus supplement and the accompanying prospectus will be made by any method that is deemed to be an “at the market offering” as defined in Rule 415(a)(4) under the Securities Act.
Each time we wish to issue and sell shares of common stock under the sales agreement, we will notify Jefferies of the number of shares to be issued, the dates on which such sales are anticipated to be made, any limitation on the number of shares to be sold in any one day and any minimum price below which sales may not be made. Once we have so instructed Jefferies, unless Jefferies declines to accept the terms of such notice, Jefferies has agreed to use its commercially reasonable efforts consistent with its normal trading and sales practices to sell such shares up to the amount specified on such terms. The obligations of Jefferies under the sales agreement to sell our shares of common stock are subject to a number of conditions that we must meet.
The settlement of sales of shares between us and Jefferies is generally anticipated to occur on the second trading day following the date on which the sale was made. Sales of our shares of common stock as contemplated in this prospectus supplement will be settled through the facilities of The Depository Trust Company or by such other means as we and Jefferies may agree upon. There is no arrangement for funds to be received in an escrow, trust or similar arrangement.
We will pay Jefferies a commission equal to 3.0% of the aggregate gross proceeds we receive from each sale of our shares of common stock. Because there is no minimum offering amount required as a condition to close this offering, the actual total public offering amount, commissions and proceeds to us, if any, are not determinable at this time. In addition, we have agreed to reimburse Jefferies for the fees and disbursements of its counsel, payable upon execution of the sales agreement, in an amount not to exceed $100,000. We estimate that the total expenses for the offering, excluding any commissions or expense reimbursement payable to Jefferies under the terms of the sales agreement, will be approximately $200,000. The remaining sale proceeds, after deducting any other transaction fees, will equal our net proceeds from the sale of such shares.
Jefferies will provide written confirmation to us before the open on The Nasdaq Capital Market on the day following each day on which shares of common stock are sold under the sales agreement. Each confirmation will include the number of shares sold on that day, the aggregate gross proceeds of such sales and the proceeds to us.
In connection with the sale of the shares of common stock on our behalf, Jefferies will be deemed to be an “underwriter” within the meaning of the Securities Act, and the compensation of Jefferies will be deemed to be underwriting commissions or discounts. We have agreed to indemnify Jefferies against certain civil liabilities, including liabilities under the Securities Act. We have also agreed to contribute to payments Jefferies may be required to make in respect of such liabilities.
The offering of our shares of common stock pursuant to the sales agreement will terminate upon the earlier of (i) the sale of all shares of common stock subject to the sales agreement and (ii) the termination of the sales agreement as permitted therein. We and Jefferies may each terminate the sales agreement at any time upon ten days’ prior notice.
This summary of the material provisions of the sales agreement does not purport to be a complete statement of its terms and conditions. A copy of the sales agreement will be filed as an exhibit to a current report on Form 8-K filed under the Exchange Act, and incorporated by reference in this prospectus supplement.
Jefferies and its affiliates may in the future provide various investment banking, commercial banking, financial advisory and other financial services for us and our affiliates, for which services they may in the future receive customary fees. In the course of its business, Jefferies may actively trade our securities for its own account or for the accounts of customers, and, accordingly, Jefferies may at any time hold long or short positions in such securities.
A prospectus supplement and the accompanying prospectus in electronic format may be made available on a website maintained by Jefferies, and Jefferies may distribute the prospectus supplement and the accompanying prospectus electronically.
S-6
The validity of the shares of common stock offered hereby will be passed upon for us by Goodwin Procter LLP, New York, New York. Jefferies LLC is being represented in connection with this offering by Cooley LLP, New York, New York.
RBSM LLP, an independent registered public accounting firm, has audited our consolidated financial statements included in our Annual Report on Form 10-K for the year ended December 31, 2018, as set forth in their report dated March 26, 2019 (which contains an explanatory paragraph about our ability to continue as a going concern), which is incorporated by reference in this prospectus supplement and elsewhere in the registration statement to which this prospectus supplement relates. Our consolidated financial statements are incorporated by reference in reliance on RBSM LLP’s report, given on their authority as experts in accounting and auditing.
WHERE YOU CAN FIND MORE INFORMATION
We file annual, quarterly and current reports, proxy statements and other information with the SEC. Our SEC filings are available to the public over the Internet at the SEC’s website at www.sec.gov. Copies of certain information filed by us with the SEC are also available on our website at www.avalon-globocare.com. Our website is not a part of this prospectus supplement and is not incorporated by reference in this prospectus supplement.
This prospectus supplement is part of a registration statement that we filed with the SEC. The registration statement contains more information than this prospectus supplement and the accompanying prospectus regarding us and the securities, including certain exhibits and schedules. You can obtain a copy of the registration statement from the SEC’s website.
The SEC allows us to incorporate by reference in this prospectus supplement and the accompanying prospectus much of the information we file with the SEC, which means that we can disclose important information to you by referring you to those publicly available documents. The information that we incorporate by reference in this prospectus supplement and the accompanying prospectus is considered to be part of this prospectus supplement and the accompanying prospectus. Because we are incorporating by reference future filings with the SEC, this prospectus supplement and the accompanying prospectus is continually updated and those future filings may modify or supersede some of the information included or incorporated in this prospectus supplement and the accompanying prospectus. This means that you must look at all of the SEC filings that we incorporate by reference to determine if any of the statements in this prospectus supplement, the accompanying prospectus or in any document previously incorporated by reference have been modified or superseded. This prospectus supplement and the accompanying prospectus incorporate by reference the documents listed below (File No. 001-38728) and any future filings we make with the SEC under Sections 13(a), 13(c), 14 or 15(d) of the Exchange Act (in each case, other than those documents or the portions of those documents not deemed to be filed) until the offering of the securities under the registration statement is terminated or completed:
| ● | Annual Report on Form 10-K for the fiscal year ended December 31, 2018, filed on March 26, 2019; |
| ● | Quarterly Reports on Form 10-Q for the fiscal quarter ended March 31, 2019, filed on May 14, 2019; for the fiscal quarter ended June 30, 2019, filed on August 15, 2019; and for the fiscal quarter ended September 30, 2019, filed on November 14, 2019 (including Amendment No. 1 to Quarterly Report on Form 10-Q filed on November 14, 2019); |
| ● | Current Reports on Form 8-K filed on January 4, 2019, January 31, 2019, March 22, 2019, April 8, 2019, April 24, 2019, April 24, 2019, April 26, 2019, August 7, 2019, September 3, 2019, September 26, 2019, October 21, 2019, October 29, 2019, October 31, 2019, November 6, 2019, November 18, 2019 and December 12, 2019; and |
| ● | The description of our common stock contained in our Registration Statement on Form 8-A filed with the SEC on November 2, 2018, including any amendments or reports filed for the purpose of updating that description. |
You may request a copy of these filings, at no cost, by writing or telephoning us at the following address or phone number:
Avalon GloboCare Corp.
4400 Route 9 South
Suite 3100
Freehold, New Jersey 07728
Attention: Chief Financial Officer
732-780-4400
S-7
PROSPECTUS

$50,000,000
Common Stock
Preferred Stock
Warrants
Units
We may offer, from time to time, in one or more offerings, common stock, preferred stock, warrants or units, which we collectively refer to as the “securities”. The aggregate initial offering price of the securities that we may offer and sell under this prospectus will not exceed $50,000,000. We may offer and sell any combination of the securities described in this prospectus in different series, at times, in amounts, at prices and on terms to be determined at, or prior to, the time of each offering. This prospectus describes the general terms of these securities and the general manner in which these securities will be offered. We will provide the specific terms of these securities in supplements to this prospectus. The prospectus supplements will also describe the specific manner in which these securities will be offered and may also supplement, update or amend information contained in this prospectus. This prospectus may not be used to consummate a sale of securities unless accompanied by the applicable prospectus supplement. You should read this prospectus and any applicable prospectus supplement before you invest.
The securities covered by this prospectus may be offered through one or more underwriters, dealers and agents or directly to purchasers. The names of any underwriters, dealers or agents, if any, will be included in a supplement to this prospectus. For general information about the distribution of securities offered, please see “Plan of Distribution”.
Our common stock is listed on the Nasdaq Capital Market under the symbol “AVCO”. On January 11, 2019, the closing price of our common stock as reported by the Nasdaq Capital Market was $3.38 per share.
We completed a 1:4 reverse stock split of its common stock on October 18, 2016. All share and per share information has been retroactively adjusted to reflect this reverse stock split.
We are an “emerging growth company” as defined in section 3(a) of the Securities Exchange Act of 1934, as amended (the “Exchange Act”), and are therefore eligible for certain exemptions from various reporting requirements applicable to reporting companies under the Exchange Act. (See “Exemptions Under the Jumpstart Our Business Startups Act.”)
Unless otherwise specified in an applicable prospectus supplement, our preferred stock, warrants and units will not be listed on any securities or stock exchange or on any automated dealer quotation system.
In reviewing this prospectus and the documents incorporated herein by reference you should carefully consider the matters described under the caption “Risk Factors”.
This investment involves a high degree of risk. You should purchase securities only if you can afford a complete loss.
Neither the Securities and Exchange Commission nor any state securities commission has approved or disapproved of these securities or determined if this prospectus is truthful or complete. Any representation to the contrary is a criminal offense.
The date of this Prospectus is February 4, 2019
TABLE OF CONTENTS
i
This prospectus is a part of a registration statement that we have filed with the SEC utilizing a “shelf” registration process. Under this shelf registration process, we may sell any combination of the securities described in this prospectus in one or more offerings up to an aggregate offering price of $50,000,000.
Each time we sell securities, we will provide a supplement to this prospectus that contains specific information about the securities being offered and the specific terms of that offering. The supplement may also add, update or change information contained in this prospectus. If there is any inconsistency between the information in this prospectus and any prospectus supplement, you should rely on the prospectus supplement.
We may offer and sell securities to, or through, underwriting syndicates or dealers, through agents or directly to purchasers. The prospectus supplement for each offering of securities will describe in detail the plan of distribution for that offering.
In connection with any offering of securities (unless otherwise specified in a prospectus supplement), the underwriters or agents may over-allot or effect transactions which stabilize or maintain the market price of the securities offered at a higher level than that which might exist in the open market. Such transactions, if commenced, may be interrupted or discontinued at any time. See “Plan of Distribution.”
Please carefully read both this prospectus and any prospectus supplement together with the documents incorporated herein by reference under “Incorporation by Reference” and the additional information described below under “Where You Can Find More Information.”
Prospective investors should be aware that the acquisition of the securities described herein may have tax consequences. You should read the tax discussion contained in the applicable prospectus supplement and consult your tax advisor with respect to your own particular circumstances.
You should rely only on the information contained or incorporated by reference in this prospectus and any prospectus supplement. We have not authorized anyone to provide you with different information. The distribution or possession of this prospectus in or from certain jurisdictions may be restricted by law. This prospectus is not an offer to sell these securities and is not soliciting an offer to buy these securities in any jurisdiction where the offer or sale is not permitted or where the person making the offer or sale is not qualified to do so or to any person to whom it is not permitted to make such offer or sale. The information contained in this prospectus is accurate only as of the date of this prospectus and any information incorporated by reference is accurate as of the date of the applicable document incorporated by reference, regardless of the time of delivery of this prospectus or of any sale of the securities. Our business, financial condition, results of operations and prospects may have changed since those dates.
In this prospectus and in any prospectus supplement, unless the context otherwise requires, references to:
| ● | the term(s) “we”, “us”, “our”, “Company”, “our company”, “Avalon GloboCare” and “Avalon” refer to Avalon GloboCare Corp., a Delaware corporation, either alone or together with our consolidated subsidiaries as the context requires. |
| ● | “Exchange Act” refers to the Securities Exchange Act of 1934, as amended. |
| ● | “Securities Act” refers to the Securities Act of 1933, as amended. |
| ● | “FINRA” refers to the Financial Industry Regulatory Authority. |
| ● | “Nasdaq” refers to the Nasdaq Capital Market. |
| ● | “SEC” or the “Commission” refers to the United States Securities and Exchange Commission. |
| ● | “prospectus” includes this document and any information incorporated herein by reference. |
We completed a 1:4 reverse stock split of its common stock on October 18, 2016. All share and per share information has been retroactively adjusted to reflect this reverse stock split.
1
Overview
We are dedicated to integrating and managing global healthcare services and resources, as well as empowering high-impact biomedical innovations and technologies to accelerate their clinical applications. Operating through two major platforms, namely “Avalon Cell” and “Avalon Rehab”, our “Technology + Service” ecosystem covers the areas of regenerative medicine, cell-based immunotherapy, exosome technology, as well as rehabilitation medicine. We plan to integrate these services through joint ventures and accretive acquisitions that bring shareholder value both in the short term, through operational entities as part of Avalon Rehab, and long term, through biomedical innovation development as part of Avalon Cell, such as our recent joint venture for the advancement of exosome isolation systems and related products.
In addition, we are engaged in the development of exosome technology to improve the diagnosis and management of diseases. Exosomes are tiny, subcellular, membrane-bound vesicles 30-150 nm in diameter that are released by almost all cell types and can carry membrane and cellular proteins, as well as genetic materials that are representative of the cell of origin. Profiling various bio-molecules in exosomes may serve as useful biomarkers for a wide variety of diseases. Our isolation system is designed to be used by researchers for biomarker discovery and clinical diagnostic development, and advancement of targeted therapies. Currently, isolation systems and service are available to isolate exosomes or extract exosomal RNA/protein from serum/plasma, urine and saliva samples. We are seeking to decode proteomic and genomic alterations underlying a wide-range of pathologies, thus allowing for the introduction of novel non-invasive “liquid biopsies”. Our mission is focused on diagnostic advancements in the fields of oncology, infectious diseases and fibrotic diseases, and the discovery of disease-specific exosomes to provide the disease origin insight necessary to enable personalized clinical management. There is no guarantee that we will be able to successfully achieve our stated mission.
We currently generate revenue by selling exosome isolation systems in China and the United States through our joint venture GenExosome Technologies, Inc. In addition, we provide medical related consulting services in advanced areas of immunotherapy and second opinion/referral services through our wholly-owned subsidiary Avalon (Shanghai) Healthcare Technology Co., Ltd., or Avalon Shanghai. We also own and operate commercial real estate in New Jersey, where we are headquartered.
On May 29, 2018, Avalon Shanghai entered into a Joint Venture Agreement with Jiangsu Unicorn Biological Technology Co., Ltd., or Unicorn, pursuant to which a company named Epicon Biosciences Co., Ltd. (“Epicon”) was formed on August 14, 2018. Epicon is owned 60% by Unicorn and 40% by Avalon Shanghai. Within two years of execution of the Joint Venture Agreement, Unicorn shall invest cash into Epicon in an amount not less than RMB 8,000,000 (approximately $1.2 million) and the premises of the laboratories of Nanjing Hospital of Chinese Medicine for exclusive use by Epicon, and Avalon Shanghai shall invest cash into Epicon in an amount not less than RMB 10,000,000 (approximately $1.5 million). As of the date of this prospectus, Unicorn has invested the premises of the laboratories of Nanjing Hospital of Chinese Medicine and Avalon Shanghai has contributed RMB 3,000,000 (approximately $0.4 million). Epicon is focused on cell preparation, third party testing, biological sample repository for commercial and scientific research purposes and the clinical transformation of scientific achievements.
On July 18, 2018, we formed a wholly owned subsidiary, Avactis Biosciences, Inc., a Nevada corporation, which will be focused on accelerating commercial activities related to Chimeric Antigen Receptor (CAR)-T technologies. The subsidiary is designed to integrate and optimize our global scientific and clinical resources to further advance the use of CAR-T to treat certain cancers.
2
On July 30, 2018, we signed a Letter of Intent with Arbele Limited, a Hong Kong company (“Arbele”) for a proposed strategic partnership agreement. The purpose of the proposed transaction is to form a joint venture company, AVAR BioTherapeutics (China) Co. Ltd., to develop, manufacture, and commercializing CAR-T immunotherapy for treating cancer patients in China, utilizing intellectual property from Arbele and the clinical platform of the LuDaopei Medical Group in China. We paid a $100,000 fee to Arbele for a five-month exclusive right to complete the definitive agreements for the transaction.
On August 6, 2018, we entered into a strategic partnership agreement with Weill Cornell’s cGMP Cellular Therapy Facility and Laboratory for Advanced Cellular Engineering headed by Dr. Yen-Michael Hsu. This strategic partnership aims to co-develop bio-production and standardization procedures in procurement, storage, processing, clinical study protocols, and bio-banking for Chimeric Antigen Receptor (CAR)-T therapy, in accordance with the Foundation of Accreditation for Cellular Therapy (FACT) and American Association of Blood Banks (AABB) standards. This partnership also includes a CAR-T education program to support and foster collaborative research and training programs for scientists and clinicians between Weill Cornell and Hebei Yanda LuDaopei Hospital, which is our main affiliated clinical facility as well as the world’s single largest medical institution in CAR-T therapy.
On October 23, 2018, Avactis Biosciences, Inc. (“Avactis”) and Arbele Limited (“Arbele”) agreed to the establishment of AVAR BioTherapeutics (China) Co. Ltd. (“AVAR”), a Sino-foreign equity joint venture, pursuant to an Equity Joint Venture Agreement (the “AVAR Agreement”), which will be owned 60% by Avactis and 40% by Arbele. The purpose and business scope of the Joint Venture is to research, develop, produce, sell, distribute and generally commercialize CAR-T/CAR-NK/TCR-T/universal cellular immunotherapy in China. Avactis is required to contribute USD $10 million (or equivalent in RMB) in cash and/or services, which shall be contributed in tranches based on milestones to be determined jointly by AVAR and Avactis in writing subject to Avactis’ cash reserves. Within 30 days, Arbele shall make contribution of USD $6.66 million in the form of entering into a License Agreement with AVAR granting AVAR with an exclusive right and license in China to its technology and intellectual property pertaining to CAR-T/CAR-NK/TCR-T/universal cellular immunotherapy technology and any additional technology developed in the future with terms and conditions to be mutually agreed upon Avactis and AVAR and services. As of the date of this prospectus, AVAR is in process of being established and the License Agreement has not been finalized.
Corporate Information
We were incorporated under the laws of the State of Delaware on July 28, 2014 under the name Global Technologies Corp. On October 18, 2016, we changed our name to Avalon GloboCare Corp. and completed a reverse split of our shares of common stock at a ratio of 1:4.
We own 100% of the capital stock of Avalon Healthcare Systems, Inc., a Delaware corporation, or AHS, which we acquired on October 19, 2016. AHS was incorporated on May 18, 2015 under the laws of the State of Delaware. In addition, we own through AHS 100% of the capital stock of Avalon (Shanghai) Healthcare Technology Co., Ltd., or Avalon Shanghai, which is a wholly foreign-owned enterprise, or WOFE, organized under the laws of the People’s Republic of China, or PRC or China. Avalon Shanghai was incorporated on April 29, 2016 and is engaged in medical related consulting services for customers. On January 23, 2017, we incorporated Avalon (BVI) Ltd, a British Virgin Islands company (dormant and in process of being dissolved). On February 7, 2017, we formed Avalon RT 9 Properties, LLC, a New Jersey limited liability company. In July 2017, we formed GenExosome Technologies Inc., a Nevada corporation, or GenExosome. On October 25, 2017, we and GenExosome entered into a Securities Purchase Agreement pursuant to which we acquired 600 shares of GenExosome in consideration of $1,326,087 in cash and 500,000 shares of our common stock. On October 25, 2017, GenExosome entered into and closed an Asset Purchase Agreement with Yu Zhou, MD, PhD, pursuant to which we acquired all assets, including all intellectual property, held by Dr. Zhou pertaining to the business of researching, developing and commercializing exosome technologies in consideration of $876,087 in cash, 500,000 shares of our common stock and 400 shares of common stock of GenExosome. As a result of the above transactions, we hold 60% of GenExosome and Dr. Zhou holds 40% of GenExosome. On October 25, 2017, GenExosome entered into and closed a Stock Purchase Agreement with Beijing Jieteng (GenExosome) Biotech Co. Ltd., a corporation incorporated in the People’s Republic of China, Beijing GenExosome, and Dr. Zhou, the sole shareholder of Beijing GenExosome, pursuant to which GenExosome acquired all of the issued and outstanding securities of Beijing GenExosome in consideration of a cash payment in the amount of $450,000.
3
On July 18, 2018, we formed a wholly owned subsidiary, Avactis Biosciences Inc. (“Avactis”), a Nevada corporation, which will be focused on accelerating commercial activities related to cellular therapies, including regenerative medicine with stem/progenitor cells as well as cellular immunotherapy including CAR-T, CAR-NK, TCR-T and others. The subsidiary is designed to integrate and optimize our global scientific and clinical resources to further advance the use of cellular therapies to treat certain cancers. On October 23, 2018, Avactis and Arbele Limited (“Arbele”) agreed to the establishment of AVAR BioTherapeutics (China) Co. Ltd. (“AVAR”), a Sino-foreign equity joint venture, pursuant to an Equity Joint Venture Agreement (the “AVAR Agreement”), which will be owned 60% by Avactis and 40% by Arbele. The purpose and business scope of the Joint Venture is to research, develop, produce, sell, distribute and generally commercialize CAR-T/CAR-NK/TCR-T/universal cellular immunotherapy in China. Avactis is required to contribute USD $10 million (or equivalent in RMB) in cash and/or services, which shall be contributed in tranches based on milestones to be determined jointly by AVAR and Avactis in writing subject to Avactis’ cash reserves. Within 30 days, Arbele shall make contribution of USD $6.66 million in the form of entering into a License Agreement with AVAR granting AVAR with an exclusive right and license in China to its technology and intellectual property pertaining to CAR-T/CAR-NK/TCR-T/universal cellular immunotherapy technology and any additional technology developed in the future with terms and conditions to be mutually agreed upon Avactis and AVAR and services. As of the date of this prospectus, AVAR is in process of being established and the License Agreement has not been finalized.
The following diagram illustrates our corporate structure as of the date of this prospectus:
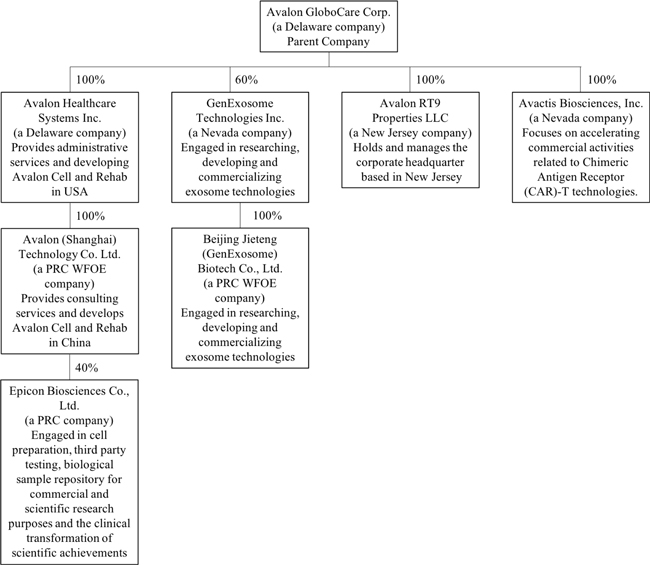
Sales and Marketing
We seek to develop new business through relationships driven by our senior management, which have extensive contacts throughout the healthcare system. Our senior management will be seeking opportunities for joint ventures, strategic relationships and acquisitions in consulting, biomedical innovations, and telemedicine, and rehabilitation centers.
4
Services
We currently generate revenue from related party strategic relationships through Avalon Shanghai that provide consultative services in advanced areas of immunotherapy and second opinion/referral services. In addition, our services are targeted at serving our clients and using our insights and deep expertise to produce tangible and significant results. Our services include research studies, executive education, daily online executive briefings, tailored expert advisory services, and consulting and management services. We typically charge an annual fee. Through our services, we attempt to have our clients focus on important problems by providing an analysis of the evolving healthcare industry and the methods prevalent in the industry to solve those problems through counsel, business planning and support. We tailor these solutions to the client’s specific strategic challenges, operational issues, and management concerns. We plan to expand our business services throughout the United States via our two major “Technology + Service” platforms: “Avalon Cell” and “Avalon Rehab”.
Strategic Partnerships
We are actively seeking potential strategic partnerships in our area of focus. In addition, we are actively seeking target acquisitions that add accretive value to our strategic plan. There is no guarantee that we will be able to successfully sign a definitive agreement, close or implement such business arrangement. Through our recent joint venture in the area of exosome technology, we are actively developing strategic relationships for the distribution and sale of our exosome isolation system and for the commercialization of exosome related products and diagnostic services.
Markets
We will focus on the following markets in developing our core business:
Platform “Avalon Cell”
Regarded as the future of medicine, we believe cell-based therapeutics will replace pharmaceuticals as a more effective and functional modality in disease treatment. We are actively engaging in this revolutionary trend and positioning to take a leading role in cell-based technology and therapeutics. The business model for our “Avalon Cell” platform is based on stringent criteria in the selection and evaluation of candidate projects at different stages of their developmental cycle. We particularly focus on projects that have strong intellectual property and distinctive innovation, as well as being translational, application-driven, and commercialization-ready. Our technology-based platform, “Avalon Cell”, comprises four programs:
| ● | Exosome technology, small extracellular vesicles that have great potential to be used as a vehicle for drug delivery in the treatment of various diseases and biomarkers for early stage diagnosis. We have commenced developing collaborative sites at Weill Cornell Medical College, MD Anderson Cancer Center and Mayo Clinic in the United States, as well as Lu Daopei Hospital of Daopei Medical Group and Da An Gene Co, Ltd., in China, focusing on exosome-based diagnostics, therapeutics, bio-banking, as well as “Exosomics Big Data”, in the unmet areas of oral cancer, ovary cancer and liver fibrosis; |
| ● | Endothelial cells, namely therapeutics involving the cells that line blood vessels and regulate exchanges between the bloodstream and surrounding tissue. These programs will occur with our collaborative sites at Weill Cornell Medical College Department of Pathology and Ansary Stem Cell Institute, focusing on standardization of endothelial cell banking and therapeutics; |
| ● | Regenerative medicine; and |
| ● | Cell-based immunotherapy (including cells such as NK, DC-CIK, CAR-T). |
5
Platform “Avalon Rehab”
A growing trend in China is in the sector of rehabilitation medicine. With our strong capabilities in integrating global technology and resources in physical medicine and rehabilitation, we will work towards positioning ourselves to take a leading role in this area through our “Avalon Rehab” platform. Our goal with this platform is to provide a turnkey, full suite of rehab services including physical therapy, occupational therapy, robotic engineering, cybernetics, and clinical nutrition. We will also engage in strategic partnerships with our institutional clients, building the leading and most authoritative network of integrated physical medicine and rehabilitation, particularly for cancer rehab patients. The focus will be on accretive acquisitions and joint venture strategic partnerships that are in revenue generating, cash flow positive positions to support biomedical innovation development while providing immediate shareholder value.
Revenue
GenExosome Technologies, Inc.
Through our majority-owned subsidiary, GenExosome Technologies, Inc., or GenExosome, we market and sell our proprietary exosome isolation systems. Exosomes are small extracellular vesicles that we believe may be used as a vehicle for drug delivery in the treatment of various diseases, and biomarkers for early stage diagnosis and as enhancements to certain cosmetic treatments and procedures. We currently produce our isolation systems in China and the U.S. and sell these systems primarily to research laboratories and universities.
Further, we generate revenue by performing development services for hospitals and sales of related products developed to hospitals through GenExosome and Beijing Jieteng (GenExosome) Biotech Co., Ltd., or Beijing GenExosome, GenExosome’s wholly-owned subsidiary.
Avalon RT 9 Properties, LLC
In May 2017, we acquired commercial property located in Freehold, New Jersey. This property is now our corporate headquarters and contains several commercial tenants that generate revenue through rental income. The revenue generated from the commercial tenants in our Freehold, New Jersey headquarters is facilitated through a management agreement with a company, which is controlled by Wenzhao Lu, our major shareholder and chairman of the Board of Directors, based in the United States.
Avalon Shanghai
We currently generate revenue by providing medical related consulting services in advanced areas of immunotherapy and second opinion/referral services through Avalon (Shanghai) Healthcare Technology Co., Ltd., or Avalon Shanghai. Our medical related consulting services include research studies, executive education, daily online executive briefings, tailored expert advisory services, and consulting and management services. Through our services we attempt to have our clients focus on important problems by providing an analysis of the evolving healthcare industry and the methods prevalent in the industry to solve those problems through counsel, business planning and support. The revenue generated from our related parties in China is managed by our employees residing in China and contactors who are retained as needed. Our contracts with the Ludaopei Hematology Research Institute Co., Ltd, a subsidiary of the Daopei Hospital Group (a related party of ours), expired as of March 31, 2018. On April 1, 2018, Avalon Shanghai entered into an advisory service contract with Beijing Ludaopei Blood Disease Research Institute Co., Ltd., a subsidiary of the Daopei Hospital Group (a related party of ours). Under the terms of the contract, the aggregate amount of advisory service fees was $300,000, which was invoiced by the end of 2018. The contract expired on December 31, 2018. Consulting services have been provided by Avalon Shanghai under the contract include:
| ● | providing scientific research consulting services; |
| ● | integrating experts, medical institutions and other resources in the United States in support of scientific research; |
6
| ● | providing technical education and training; and |
| ● | assisting in publication of academic papers. |
Strategic Development
We intend to focus on three components. The initial component will be focused on acquiring and/or managing fixed assets including healthcare real estate as well as stem cell banks. In addition, we intend to pursue the acquisition and development of healthcare related technologies for cell related diagnostics and therapeutics through acquisition, licensing or joint ventures with major universities and biotech companies. We will also consider a third avenue of investing in certain technologies for cell related diagnostics and therapeutics.
Intellectual Property
Our goal is to obtain, maintain and enforce patent rights for our products, formulations, processes, methods of use and other proprietary technologies, preserve our trade secrets, and operate without infringing on the proprietary rights of other parties, both in the United States and abroad. Our policy is to actively seek to obtain, where appropriate, the broadest intellectual property protection possible for our current product candidates and any future product candidates, proprietary information and proprietary technology through a combination of contractual arrangements and patents, both in the United States and abroad. Even patent protection, however, may not always afford us with complete protection against competitors who seek to circumvent our patents. If we fail to adequately protect or enforce our intellectual property rights or secure rights to patents of others, the value of our intellectual property rights would diminish. To this end, we require all of our employees, consultants, advisors and other contractors to enter into confidentiality agreements that prohibit the disclosure and use of confidential information and, where applicable, require disclosure and assignment to us of the ideas, developments, discoveries and inventions relevant to our technologies and important to our business.
Through GenExosome, we have applied for four patents in China with related trademarks. We are in the process of applying for those same patents and trademarks in the United States and are also in the process of developing additional patents and related intellectual property. We own and control a variety of trade secrets, confidential information, trademarks, trade names, copyrights, and other intellectual property rights that, in the aggregate, are of material importance to our business. We consider our trademarks, service marks, and other intellectual property to be proprietary, and rely on a combination of copyright, trademark, trade secret, non-disclosure, and contractual safeguards to protect our intellectual property rights.
Current patent applications in China are as follows.
| Application of an Exosomal MicroRNA in plasma as biomarker to diagnosis LIVER CANCER | Patent
application number: CN 2016 1 0675107.5 | |
| Clinical application of circulating exosome carried miRNA-33b in the diagnosis of liver cancer | Patent application
number: CN 2016 1 0675110.7 | |
| Saliva exosome-based methods and composition for the Diagnosis, Staging and Prognosis of ORAL CANCER | Patent application
number: CN 2017 1 0330847.X | |
| A novel exosome-based therapeutics against proliferative oral diseases | Patent application
number: CN 2017 1 0330835.7 |
Competition
GenExosome Technologies, Inc.
We currently market for sale our proprietary exosome isolation system. There are other companies that produce exosome isolation systems. However, our internal analysis shows that most exosome isolation systems use a centrifuge process for isolation which takes several hours and results in a low purity. Our isolation system is a membrane system which isolates exosomes in a few minutes with a higher purity than competing systems.
7
We believe that our proprietary isolation system is superior to competing systems and plan to continue to improve our process to maintain competitive advantages in the market.
Avalon Shanghai
In our current consulting business in the People’s Republic of China, or PRC or China, we compete with a number of advisory firm offering similar service including consulting and strategy firms; market research, data, benchmarking, and forecasting providers; technology vendors and services firms; healthcare information technology firms; technology advisory firms; outsourcing firms; and specialized providers of educational and training services. Other organizations, such as state and national trade associations, group purchasing organizations, non-profit think-tanks, and database companies, also may offer research, consulting, tools, and education services to health care and education organizations.
We believe that the principal competitive factors in our market include quality and timeliness of our services, strength and depth of relationships with our clients, ability to meet the changing needs of current and prospective clients, measurable returns on customer investment, and service and affordability.
As our business develops and we expand through joint ventures, acquisitions and strategic partnerships in the U.S. and PRC, we will have competition with other direct service providers, emerging technologies and medical communication platforms. We will seek to maintain a competitive advantage through intellectual property, superior quality management and cutting edge technology.
Rt. 9 Properties, LLC
Our executive commercial building in Freehold, New Jersey is located on a major highway and is one of the largest buildings in the surrounding areas. It is centrally located and maintains high occupancy. There are other commercial properties in the vicinity that offer similar amenities. However, premier executive offices are limited and as such we expect to continue to maintain high occupancy in the near term.
Manufacturing
GenExosome presently maintains its laboratory, research and manufacturing facilities in leased premises located in Beijing, China and Columbus, Ohio. We manufacture and assemble our exosome isolation systems for sale to research laboratories and universities. The exosome isolation system is comprised of our proprietary reagent with specifically designed membranes. We assemble the isolation system at our premises through commercially available purchased components that we modify in a proprietary manner and assemble in our systems, which are then shipped to our customers.
Legal Proceedings
From time to time, we are subject to ordinary routine litigation incidental to our normal business operations. We are not currently a party to, and our property is not subject to, any material legal proceedings.
Properties
Our principal offices are located at 4400 Route 9 South, Freehold, NJ 07728. The office building is owned by our subsidiary, Avalon RT 9 Properties, LLC, which is in business of owning and operating an income-producing real property. Our property is well maintained, adequately meets our needs, and is being utilized for its intended purpose.
We lease additional office space for operations. Office location is not crucial to our operations, and we anticipate no difficulty in extending these leases or obtaining comparable office space.
We are obligated under various lease agreements providing for office space that expire at various dates through the year 2019. Total rent expense under these lease agreements was $138,307 and $2,000 for the years ended December 31, 2017 and 2016, respectively.
8
We believe that our current office space is adequate for our current and immediately foreseeable operating needs.
Employees
As of December 20, 2018, we had 11 employees, nine of which are full time employees. Three full time employees and one part time employee are in the U.S. and six full time and one part time employees are in China. None of our employees are represented by a collective bargaining arrangement.
Government Regulation
Overview
The healthcare industry in the PRC and U.S. is highly regulated and subject to changing political, legislative, regulatory, and other influences. Further, the healthcare industry is currently undergoing rapid change. We are uncertain how, when or in what context these new changes will be adopted or implemented. These new regulations could create unexpected liabilities for us, could cause us or our members to incur additional costs and could restrict our or our clients’ operations. Many of the laws are complex and their application to us, our clients, or the specific services and relationships we have with our members are not always clear. Our failure to anticipate accurately the application of these laws and regulations, or our other failure to comply, could create liability for us, result in adverse publicity, and otherwise negatively affect our business.
Despite efforts to develop its legal system over the past several decades, including but not limited to legislation dealing with economic matters such as foreign investment, corporate organization and governance, commerce, taxation and trade, the PRC continues to lack a comprehensive system of laws. Further, the laws that do exist in the PRC are often vague, ambiguous and difficult to enforce, which could negatively affect our ability to do business in China and compete with other companies in our segments.
In September 2006, the Ministry of Commerce, or MOFCOM, promulgated the Regulations on Foreign Investors’ Mergers and Acquisitions of Domestic Enterprises, or the M&A Regulations, in an effort to better regulate foreign investment in the PRC. The M&A Regulations were adopted in part as a needed codification of certain joint venture formation and operating practices, and also in response to the government’s increasing concern about protecting domestic companies in perceived key industries and those associated with national security, as well as the outflow of well-known trademarks, including traditional Chinese brands.
As a U.S. based company doing business in the PRC, we seek to comply with all PRC laws, rules and regulations and pronouncements, and endeavor to obtain all necessary approvals from applicable PRC regulatory agencies such as the MOFCOM, the State Assets Supervision and Administration Commission, the State Administration for Taxation, the State Administration for Industry and Commerce, the China Securities Regulatory Commission, and the State Administration of Foreign Exchange, or SAFE.
Drug Approval Process
The research, development, testing, manufacture, labeling, promotion, advertising, distribution and marketing, among other things, of our product candidates are extensively regulated by governmental authorities in the United States and other countries. In the United States, the FDA regulates drugs under the Federal Food, Drug, and Cosmetic Act, or the FDCA, and its implementing regulations. Failure to comply with the applicable U.S. requirements may subject us to administrative or judicial sanctions, such as the FDA’s refusal to approve a pending new drug application, or NDA, or a pending biologics license application, or BLA, warning letters, product recalls, product seizures, total or partial suspension of production or distribution, injunctions and/or criminal prosecution.
9
Pharmaceutical products such as ours may not be commercially marketed without prior approval from the FDA and comparable regulatory agencies in other countries. In the United States, the process to receiving such approval is long, expensive and risky, and includes the following steps:
| ● | pre-clinical laboratory tests, animal studies, and formulation studies; | |
| ● | submission to the FDA of an IND for human clinical testing, which must become effective before human clinical trials may begin; | |
| ● | adequate and well-controlled human clinical trials to establish the safety and efficacy of the drug for each indication; | |
| ● | submission to the FDA of an NDA or BLA; | |
| ● | satisfactory completion of an FDA inspection of the manufacturing facility or facilities at which the drug is produced to assess compliance with current good manufacturing practices, or cGMPs; | |
| ● | a potential FDA audit of the preclinical and clinical trial sites that generated the data in support of the NDA or BLA; | |
| ● | the ability to obtain clearance or approval of companion diagnostic tests, if required, on a timely basis, or at all; and | |
| ● | FDA review and approval of the NDA or BLA. |
Regulation by U.S. and foreign governmental authorities is a significant factor affecting our ability to commercialize any of our products, as well as the timing of such commercialization and our ongoing research and development activities. The commercialization of drug products requires regulatory approval by governmental agencies prior to commercialization. Various laws and regulations govern or influence the research and development, non-clinical and clinical testing, manufacturing, processing, packing, validation, safety, labeling, storage, record keeping, registration, listing, distribution, advertising, sale, marketing and post-marketing commitments of our products. The lengthy process of seeking these approvals, and the subsequent compliance with applicable laws and regulations, require expending substantial resources.
The results of pre-clinical testing, which include laboratory evaluation of product chemistry and formulation, animal studies to assess the potential safety and efficacy of the product and its formulations, details concerning the drug manufacturing process and its controls, and a proposed clinical trial protocol and other information must be submitted to the FDA as part of an IND that must be reviewed and become effective before clinical testing can begin. The study protocol and informed consent information for patients in clinical trials must also be submitted to an independent Institutional Review Board, or IRB, for approval covering each institution at which the clinical trial will be conducted. Once a sponsor submits an IND, the sponsor must wait 30 calendar days before initiating any clinical trials. If the FDA has comments or questions within this 30-day period, the issue(s) must be resolved to the satisfaction of the FDA before clinical trials can begin. In addition, the FDA, an IRB or the company may impose a clinical hold on ongoing clinical trials due to safety concerns. If the FDA imposes a clinical hold, clinical trials can only proceed under terms authorized by the FDA. Our pre-clinical and clinical studies must conform to the FDA’s Good Laboratory Practice, or GLP, and Good Clinical Practice, or GCP, requirements, respectively, which are designed to ensure the quality and integrity of submitted data and protect the rights and well-being of study patients. Information for certain clinical trials also must be publicly disclosed within certain time limits on the clinical trial registry and results databank maintained by the NIH.
Typically, clinical testing involves a three-phase process; however, the phases may overlap or be combined:
| ● | Phase I clinical trials typically are conducted in a small number of volunteers or patients to assess the early tolerability and safety profile, and the pattern of drug absorption, distribution and metabolism; | |
| ● | Phase II clinical trials typically are conducted in a limited patient population with a specific disease in order to assess appropriate dosages and dose regimens, expand evidence of the safety profile and evaluate preliminary efficacy; and | |
| ● | Phase III clinical trials typically are larger scale, multicenter, well-controlled trials conducted on patients with a specific disease to generate enough data to statistically evaluate the efficacy and safety of the product, to establish the overall benefit-risk relationship of the drug and to provide adequate information for the registration of the drug. |
10
A therapeutic product candidate being studied in clinical trials may be made available for treatment of individual patients, in certain circumstances. Pursuant to the 21st Century Cures Act (Cures Act), which was signed into law in December 2016. The manufacturer of an investigational product for a serious disease or condition is required to make available, such as by posting on its website, its policy on evaluating and responding to requests for individual patient access to such investigational product.
The results of the pre-clinical and clinical testing, chemistry, manufacturing and control information, proposed labeling and other information are then submitted to the FDA in the form of either an NDA or BLA for review and potential approval to begin commercial sales. In responding to an NDA or BLA, the FDA may grant marketing approval, request additional information in a Complete Response Letter, or CRL, or deny the approval if it determines that the NDA or BLA does not provide an adequate basis for approval. A CRL generally contains a statement of specific conditions that must be met in order to secure final approval of an NDA or BLA and may require additional testing. If and when those conditions have been met to the FDA’s satisfaction, the FDA will typically issue an approval letter, which authorizes commercial marketing of the product with specific prescribing information for specific indications, and sometimes with specified post-marketing commitments and/or distribution and use restrictions imposed under a Risk Evaluation and Mitigation Strategy program. Any approval required from the FDA might not be obtained on a timely basis, if at all.
Among the conditions for an NDA or BLA approval is the requirement that the manufacturing operations conform on an ongoing basis with cGMPs. In complying with cGMPs, we must expend time, money and effort in the areas of training, production and quality control within our own organization and at our contract manufacturing facilities. A successful inspection of the manufacturing facility by the FDA is usually a prerequisite for final approval of a pharmaceutical product. Following approval of the NDA or BLA, we and our manufacturers will remain subject to periodic inspections by the FDA to assess compliance with cGMPs requirements and the conditions of approval. We will also face similar inspections coordinated by foreign regulatory authorities.
Disclosure of Clinical Trial Information
Sponsors of certain clinical trials of FDA-regulated products are required to register and disclose certain clinical trial information. Information related to the product, patient population, phase of investigation, trial sites and investigators, and other aspects of the clinical trial are then made public as part of the registration. Sponsors are also obligated to disclose the results of their clinical trials after completion. Disclosure of the results of these trials can be delayed in certain circumstances for up to two years after the date of completion of the trial. Competitors may use this publicly available information to gain knowledge regarding the progress of development programs.
Expedited Development and Review Programs
The FDA has a Fast Track program that is intended to expedite or facilitate the process for reviewing new drugs and biological products that meet certain criteria. Specifically, new drugs and biological products are eligible for Fast Track designation if they are intended to treat a serious or life-threatening condition and demonstrate the potential to address unmet medical needs for the condition. Fast Track designation applies to the combination of the product and the specific indication for which it is being studied. The sponsor of a new drug or biologic may request the FDA to designate the drug or biologic as a Fast Track product at any time during the clinical development of the product. Unique to a Fast Track product, the FDA may consider for review sections of the marketing application on a rolling basis before the complete application is submitted, if the sponsor provides a schedule for the submission of the sections of the application, the FDA agrees to accept sections of the application and determines that the schedule is acceptable, and the sponsor pays any required user fees upon submission of the first section of the application.
11
Any product submitted to the FDA for marketing, including under a Fast Track program, may be eligible for other types of FDA programs intended to expedite development and review, such as priority review and accelerated approval. Under the Breakthrough Therapy program, products intended to treat a serious or life-threatening disease or condition may be eligible for the benefits of the Fast Track program when preliminary clinical evidence demonstrates that such product may have substantial improvement on one or more clinically significant endpoints over existing therapies. Additionally, FDA will seek to ensure the sponsor of a breakthrough therapy product receives timely advice and interactive communications to help the sponsor design and conduct a development program as efficiently as possible. Any product is eligible for priority review if it has the potential to provide safe and effective therapy where no satisfactory alternative therapy exists or a significant improvement in the treatment, diagnosis or prevention of a disease compared to marketed products. The FDA will attempt to direct additional resources to the evaluation of an application for a new drug or biological product designated for priority review in an effort to facilitate the review. Additionally, a product may be eligible for accelerated approval. Drug or biological products studied for their safety and effectiveness in treating serious or life-threatening illnesses and that provide meaningful therapeutic benefit over existing treatments may receive accelerated approval, which means that they may be approved on the basis of adequate and well-controlled clinical studies establishing that the product has an effect on a surrogate endpoint that is reasonably likely to predict a clinical benefit, or on the basis of an effect on a clinical endpoint other than survival or irreversible morbidity. As a condition of approval, the FDA may require that a sponsor of a drug or biological product receiving accelerated approval perform adequate and well-controlled post-marketing clinical studies. In addition, the FDA currently requires as a condition for accelerated approval the pre-approval of promotional materials, which could adversely impact the timing of the commercial launch of the product. Fast Track designation, Breakthrough Therapy designation, priority review and accelerated approval do not change the standards for approval but may expedite the development or approval process.
Regenerative Medicine Advanced Therapies (RMAT) Designation
The FDA has established a Regenerative Medicine Advanced Therapy, or RMAT, designation as part of its implementation of the 21st Century Cures Act, or Cures Act. The RMAT designation program is intended to fulfill the Cures Act requirement that the FDA facilitate an efficient development program for, and expedite review of, any drug that meets the following criteria: (1) it qualifies as a RMAT, which is defined as a cell therapy, therapeutic tissue engineering product, human cell and tissue product, or any combination product using such therapies or products, with limited exceptions; (2) it is intended to treat, modify, reverse, or cure a serious or life-threatening disease or condition; and (3) preliminary clinical evidence indicates that the drug has the potential to address unmet medical needs for such a disease or condition. Like breakthrough therapy designation, RMAT designation provides potential benefits that include more frequent meetings with FDA to discuss the development plan for the product candidate, and eligibility for rolling review and priority review. Products granted RMAT designation may also be eligible for accelerated approval on the basis of a surrogate or intermediate endpoint reasonably likely to predict long-term clinical benefit, or reliance upon data obtained from a meaningful number of sites, including through expansion to additional sites. RMAT-designated products that receive accelerated approval may, as appropriate, fulfill their post-approval requirements through the submission of clinical evidence, clinical studies, patient registries, or other sources of real world evidence (such as electronic health records); through the collection of larger confirmatory data sets; or via post-approval monitoring of all patients treated with such therapy prior to approval of the therapy.
Post-Approval Requirements
Oftentimes, even after a drug has been approved by the FDA for sale, the FDA may require that certain post-approval requirements be satisfied, including the conduct of additional clinical studies. If such post-approval requirements are not satisfied, the FDA may withdraw its approval of the drug. In addition, holders of an approved NDA or BLA are required to report certain adverse reactions to the FDA, comply with certain requirements concerning advertising and promotional labeling for their products, and continue to have quality control and manufacturing procedures conform to cGMPs after approval. The FDA periodically inspects the sponsor’s records related to safety reporting and/or manufacturing facilities; this latter effort includes assessment of compliance with cGMPs. Accordingly, manufacturers must continue to expend time, money, and effort in the area of production and quality control to maintain cGMPs compliance.
Pricing, Coverage and Reimbursement
Sales of pharmaceutical products depend, in part, on the extent to which the costs of products are covered and paid for by third-party payors, such as government health programs, commercial insurance, and managed healthcare organizations. Third-party payors may limit coverage to specific products on an approved list or formulary, which might not include all of the FDA-approved products for a particular indication. Also, third-party payors may refuse to include a particular branded drug on their formularies or otherwise restrict patient access to a branded drug when a less costly generic equivalent or another alternative is available. Third-party payors are increasingly challenging the prices charged for medical products and services. Additionally, the containment of healthcare costs has become a priority of federal and state governments, and the prices of drugs have been a focus in this effort. The U.S. government, state legislatures and foreign governments have shown significant interest in implementing cost-containment programs, including price controls, restrictions on reimbursement and requirements for substitution of generic products. The current U.S. administration has indicated support for possible new measures to regulate drug pricing.
12
For example, the Patient Protection and Affordable Care Act, as amended by the Health Care and Education Affordability Reconciliation Act of 2010, collectively referred to as the ACA, enacted in March 2010, has had a significant impact on the health care industry by, for example, expanding coverage for the uninsured and seeking to contain overall healthcare costs. With regard to pharmaceutical products, among other things, the ACA contains provisions that may reduce the profitability of drug products such as expanding and increasing industry rebates for drugs covered under Medicaid programs and making changes to the coverage requirements under the Medicare Part D program. Recently, the current U.S. administration and U.S. Congress have expressed a desire to modify, repeal, or otherwise invalidate all, or certain provisions of, the ACA, which has contributed to the uncertainty of the ongoing implementation and impact of the ACA and also underscores the potential for additional health care reform going forward. For example, the newly enacted federal income tax law includes a provision repealing, effective January 1, 2019, the tax-based shared responsibility payment imposed by the ACA on certain individuals who fail to maintain qualifying health coverage for all or part of a year that is commonly referred to as the “individual mandate.” Congress may consider other legislation that would alter other aspects of the ACA. There is still uncertainty with respect to the impact the current U.S. administration and the U.S. Congress may have, if any, and any changes will likely take time to unfold.
Further other legislative changes have been proposed and adopted since the ACA was enacted. For example, in August 2011, President Obama signed into law the Budget Control Act of 2011, which, among other things, created the Joint Select Committee on Deficit Reduction to recommend to Congress proposals in spending reductions. The Joint Select Committee on Deficit Reduction did not achieve a targeted deficit reduction of at least $1.2 trillion for fiscal years 2012 through 2021, triggering the legislation’s automatic reduction to several government programs. This includes aggregate reductions to Medicare payments to providers of up to 2% per fiscal year, which went into effect beginning on April 1, 2013 and will stay in effect through 2027 unless additional Congressional action is taken. In addition, on February 9, 2018, Congress passed the Bipartisan Budget Act that made a number of healthcare reforms. For example, the law changes the discounts manufacturers are required to apply to their drugs under the Coverage Gap Discount Program from 50% to 70% of the negotiated price starting in 2019. In addition, the law increases civil and criminal penalties for fraud and abuse laws, including, for example, criminal fines for violations of the Anti-Kickback Statute increase from $25,000 to $100,000 and corresponding prison sentences also increase from no more than five years to no more than ten years.
There has also been heightened governmental scrutiny recently over the manner in which drug manufacturers set prices for their marketed products, which have resulted in several Congressional inquiries and proposed bills designed to, among other things, bring more transparency to product pricing, review the relationship between pricing and manufacturer patient programs, and reform government program reimbursement methodologies for drug products. Individual states in the United States have also become increasingly aggressive in passing legislation and implementing regulations designed to control pharmaceutical and biological product pricing, including price or patient reimbursement constraints, discounts, restrictions on certain product access and marketing cost disclosure and transparency measures. For example, in September 2017, the California State Assembly approved SB17 which requires pharmaceutical companies to notify health insurers and government health plans at least 60 days before any scheduled increases in the prices of their products if they exceed 16% over a two-year period, and further requiring pharmaceutical companies to explain the reasons for such increase.
In addition, in some non-U.S. jurisdictions, the proposed pricing for a product candidate must be approved before it may be lawfully marketed. The requirements governing drug pricing vary widely from country to country. For example, the EU provides options for its member states to restrict the range of medicinal products for which their national health insurance systems provide reimbursement and to control the prices of medicinal products for human use. A member state may approve a specific price for the medicinal product or it may instead adopt a system of direct or indirect controls on the profitability of the company placing the medicinal product on the market. There can be no assurance that any country that has price controls or reimbursement limitations for pharmaceutical products will allow favorable reimbursement and pricing arrangements for any of our product candidates. Historically, product candidates launched in the EU do not follow price structures of the U.S. and generally tend to have price structures that are significantly lower.
13
Other Healthcare Fraud and Abuse Laws
In the U.S., our activities are potentially subject to regulation by various federal, state and local authorities in addition to the FDA, including but not limited to, the Centers for Medicare and Medicaid Services, or CMS, other divisions of the U.S. Department of Health and Human Services (such as the Office of Inspector General and the Health Resources and Service Administration), the U.S. Department of Justice, or the DOJ, and individual U.S. Attorney offices within the DOJ, and state and local governments. For example, sales, marketing and scientific/educational grant programs may have to comply with the anti-fraud and abuse provisions of the Social Security Act, the false claims laws, the privacy and security provisions of the Health Insurance Portability and Accountability Act, or HIPAA, and similar state laws, each as amended, as applicable.
The federal Anti-Kickback Statute prohibits, among other things, any person or entity from knowingly and willfully offering, paying, soliciting or receiving any remuneration, directly or indirectly, overtly or covertly, in cash or in kind, to induce or in return for purchasing, leasing, ordering or arranging for the purchase, lease or order of any item or service reimbursable, in whole or in part, under Medicare, Medicaid or other federal healthcare programs. The term remuneration has been interpreted broadly to include anything of value. The Anti-Kickback Statute has been interpreted to apply to arrangements between therapeutic product manufacturers on one hand and prescribers, purchasers, and formulary managers on the other. There are a number of statutory exceptions and regulatory safe harbors protecting some common activities from prosecution. The exceptions and safe harbors are drawn narrowly and practices that involve remuneration that may be alleged to be intended to induce prescribing, purchasing or recommending may be subject to scrutiny if they do not qualify for an exception or safe harbor. Failure to meet all of the requirements of a particular applicable statutory exception or regulatory safe harbor does not make the conduct per se illegal under the Anti-Kickback Statute. Instead, the legality of the arrangement will be evaluated on a case-by-case basis based on a cumulative review of all of its facts and circumstances. Additionally, the intent standard under the Anti-Kickback Statute was amended by the ACA to a stricter standard such that a person or entity no longer needs to have actual knowledge of the statute or specific intent to violate it in order to have committed a violation. In addition, the ACA codified case law that a claim including items or services resulting from a violation of the federal Anti-Kickback Statute constitutes a false or fraudulent claim for purposes of the federal False Claims Act, or FCA.
The federal false claims and civil monetary penalty laws, including the FCA, which imposes significant penalties and can be enforced by private citizens through civil qui tam actions, prohibit any person or entity from, among other things, knowingly presenting, or causing to be presented, a false or fraudulent claim for payment to, or approval by, the federal healthcare programs, including Medicare and Medicaid, or knowingly making, using, or causing to be made or used a false record or statement material to a false or fraudulent claim to the federal government. A claim includes “any request or demand” for money or property presented to the U.S. government. For instance, historically, pharmaceutical and other healthcare companies have been prosecuted under these laws for allegedly providing free product to customers with the expectation that the customers would bill federal programs for the product. Other companies have been prosecuted for causing false claims to be submitted because of the companies’ marketing of the product for unapproved, off-label, and thus generally non-reimbursable, uses.
HIPAA created additional federal criminal statutes that prohibit, among other things, knowingly and willfully executing, or attempting to execute, a scheme to defraud or to obtain, by means of false or fraudulent pretenses, representations or promises, any money or property owned by, or under the control or custody of, any healthcare benefit program, including private third-party payors, willfully obstructing a criminal investigation of a healthcare offense, and knowingly and willfully falsifying, concealing or covering up by trick, scheme or device, a material fact or making any materially false, fictitious or fraudulent statement in connection with the delivery of or payment for healthcare benefits, items or services. Like the Anti-Kickback Statute, the ACA amended the intent standard for certain healthcare fraud statutes under HIPAA such that a person or entity no longer needs to have actual knowledge of the statute or specific intent to violate it in order to have committed a violation.
Many states have similar, and typically more prohibitive, fraud and abuse statutes or regulations that apply to items and services reimbursed under Medicaid and other state programs, or, in several states, apply regardless of the payor. Additionally, to the extent that our product candidates may in the future be sold in a foreign country, we may be subject to similar foreign laws.
14
We may be subject to data privacy and security regulations by both the federal government and the states in which we conduct our business. HIPAA, as amended by the Health Information Technology for Economic and Clinical Health Act, or HITECH, and its implementing regulations, imposes requirements relating to the privacy, security and transmission of individually identifiable health information. Among other things, HITECH makes HIPAA’s privacy and security standards directly applicable to business associates, independent contractors, or agents of covered entities that receive or obtain protected health information in connection with providing a service on behalf of a covered entity. HITECH also created four new tiers of civil monetary penalties, amended HIPAA to make civil and criminal penalties directly applicable to business associates, and gave state attorneys general new authority to file civil actions for damages or injunctions in federal courts to enforce HIPAA and seek attorneys’ fees and costs associated with pursuing federal civil actions. In addition, many state laws govern the privacy and security of health information in specified circumstances, many of which differ from each other in significant ways, are often not pre-empted by HIPAA, and may have a more prohibitive effect than HIPAA, thus complicating compliance efforts.
We expect our product, after approval, may be eligible for coverage under Medicare, the federal health care program that provides health care benefits to the aged and disabled, and covers outpatient services and supplies, including certain pharmaceutical products, that are medically necessary to treat a beneficiary’s health condition. In addition, the product may be covered and reimbursed under other government programs, such as Medicaid and the 340B Drug Pricing Program. The Medicaid Drug Rebate Program requires pharmaceutical manufacturers to enter into and have in effect a national rebate agreement with the Secretary of the Department of Health and Human Services as a condition for states to receive federal matching funds for the manufacturer’s outpatient drugs furnished to Medicaid patients. Under the 340B Drug Pricing Program, the manufacturer must extend discounts to entities that participate in the program. As part of the requirements to participate in certain government programs, many pharmaceutical manufacturers must calculate and report certain price reporting metrics to the government, such as average manufacturer price, or AMP, and best price. Penalties may apply in some cases when such metrics are not submitted accurately and timely.
Additionally, the federal Physician Payments Sunshine Act, or the Sunshine Act, within the ACA, and its implementing regulations, require that certain manufacturers of drugs, devices, biological and medical supplies for which payment is available under Medicare, Medicaid or the Children’s Health Insurance Program (with certain exceptions) report annually to CMS information related to certain payments or other transfers of value made or distributed to physicians and teaching hospitals, or to entities or individuals at the request of, or designated on behalf of, the physicians and teaching hospitals and to report annually certain ownership and investment interests held by physicians and their immediate family members. Failure to report accurately could result in penalties. In addition, many states also govern the reporting of payments or other transfers of value, many of which differ from each other in significant ways, are often not pre-empted, and may have a more prohibitive effect than the Sunshine Act, thus further complicating compliance efforts.
New Legislation and Regulations
From time to time, legislation is drafted, introduced and passed in Congress that could significantly change the statutory provisions governing the testing, approval, manufacturing and marketing of products regulated by the FDA. In addition to new legislation, FDA regulations and policies are often revised or interpreted by the agency in ways that may significantly affect our business and our products. It is impossible to predict whether further legislative changes will be enacted or whether FDA regulations, guidance, policies or interpretations will be changed or what the effect of such changes, if any, may be.
15
Company History
On October 19, 2016, we entered into and closed a Share Exchange Agreement with the shareholders of Avalon Healthcare System, Inc., a Delaware corporation, or AHS, each of which are accredited investors, or the AHS Shareholders, pursuant to which we acquired 100% of the outstanding securities of AHS in exchange for 50,000,000 shares of our common stock, or the AHS Acquisition. Considering that, following the acquisition, the AHS Shareholders control the majority of our outstanding voting common stock and we effectively succeeded our otherwise minimal operations to those that are theirs, AHS is considered the accounting acquirer in this reverse-acquisition transaction. A reverse-acquisition transaction is considered, and accounted for as, a capital transaction in substance; it is equivalent to the issuance of AHS securities for our net monetary assets, which are deminimus, accompanied by a recapitalization. Accordingly, we have not recognized any goodwill or other intangible assets in connection with this reverse acquisition transaction. AHS is the surviving and continuing entity and the historical financials following the reverse acquisition transaction will be those of AHS. We were a “shell company” (as such term is defined in Rule 12b-2 under the Securities Exchange Act of 1934, as amended) immediately prior to our acquisition of AHS pursuant to the terms of the Share Exchange Agreement. AHS owns 100% of the capital stock of Avalon (Shanghai) Healthcare Technology Co., Ltd., or Avalon Shanghai, which is a wholly foreign-owned enterprise organized under the laws of the PRC. Avalon Shanghai was incorporated on April 29, 2016 and is engaged in medical related consulting services for customers. Consequently, we believe that acquisition has caused us to cease to be a shell company as we no longer have nominal operations.
On September 29, 2016, effective October 18, 2016, we filed a Certificate of Amendment of Certificate of Incorporation, or the Certificate, with the State of Delaware to (i) effect a reverse stock split of our outstanding and authorized shares of common stock at a ratio of 1 for 4, or the Reverse Stock Split, and (ii) effectuate a name change, or the Name Change. Fractional shares that resulted from the Reverse Stock Split were rounded up to the next highest number. As a result of the Name Change, our name changed from “Global Technologies Corp.” to “Avalon GloboCare Corp.” The Certificate was approved by the majority of our shareholders and by our Board of Directors. The effective date of the Reverse Stock Split and the Name Change was October 18, 2016.
On December 22, 2016, we entered into an Agreement of Sale, or the Purchase Agreement, with Freehold Craig Road Partnership, a New Jersey partnership, to purchase certain real property located in the Township of Freehold, County of Monmouth, State of New Jersey, having a street address of 4400 Route 9 South, Freehold, NJ 07728. All rights under the Purchase Agreement were assigned by us to Avalon RT 9 Properties, LLC, our wholly-owned subsidiary, or Avalon RT 9. Avalon RT 9 closed on the purchase of the property on May 5, 2017. The purchase price including adjustments paid by us for the property was $7.65 million in cash. The seller also assigned all lease agreements for all tenants on the property to Avalon RT 9.
In July 2017, we formed GenExosome Technologies Inc., a Nevada corporation, or GenExosome. On September 29, 2017, Dr. David K. Jin was appointed as the sole director and as the Chief Executive Officer, Chief Medical Officer and President, Meng Li was appointed as Chief Operating Officer and Secretary and Luisa Ingargiola was appointed as Chief Financial Officer. On October 25, 2017, we and GenExosome entered into a Securities Purchase Agreement pursuant to which we acquired 600 shares of GenExosome in consideration of $1,326,087 in cash and 500,000 shares of our common stock.
On October 25, 2017, GenExosome entered into and closed an Asset Purchase Agreement with Yu Zhou, MD, PhD, pursuant to which we acquired all assets, including all intellectual property, held by Dr. Zhou pertaining to the business of researching, developing and commercializing exosome technologies including, but not limited to, patent application number CN 2016 1 0675107.5 (application of an Exosomal MicroRNA in plasma as biomarker to diagnosis liver cancer), patent application number CN 2016 1 0675110.7 (clinical application of circulating exosome carried miRNA-33b in the diagnosis of liver cancer), patent application number CN 2017 1 0330847.X (saliva exosome based methods and composition for the diagnosis, staging and prognosis of oral cancer) and patent application number CN 2017 1 0330835.7 (a novel exosome-based therapeutics against proliferative oral diseases). In consideration of the assets, GenExosome agreed to pay Dr. Zhou $876,087 in cash no later than November 24, 2017, transfer 500,000 shares of our common stock to Dr. Zhou no later than November 24, 2017 and issue Dr. Zhou 400 shares of common stock of GenExosome no later than November 24, 2017. The above transactions have since been completed and as a result, we hold 60% of GenExosome and Dr. Zhou holds 40% of GenExosome.
On October 25, 2017, GenExosome entered into and closed a Stock Purchase Agreement with Beijing Jieteng (GenExosome) Biotech Co. Ltd., a corporation incorporated in the People’s Republic of China, or Beijing GenExosome, and Dr. Zhou, the sole shareholder of Beijing GenExosome, pursuant to which GenExosome acquired all of the issued and outstanding securities of Beijing GenExosome in consideration of a cash payment in the amount of $450,000, which shall be paid upon Beijing GenExosome recording the change in ownership with the Ministry of Commerce of the People’s Republic of China in accordance with the Interim Measures for Record Management regarding the Establishment and Change of Foreign-invested Enterprises (revised), which we expect to be completed in the second quarter of 2018.
16
On October 25, 2017, GenExosome increased its size of its board of directors from one to four and appointed Wenzhao “Daniel” Lu, Meng Li and Dr. Zhou to the board of directors. In addition, Dr. Zhou was appointed as Co-Chief Executive Officer of GenExosome.
On October 25, 2017, Dr. Zhou and GenExosome entered into an Executive Retention Agreement pursuant to which Dr. Zhou agreed to serve as Co-Chief Executive Officer in consideration of an annual salary of $160,000. Dr. Zhou and GenExosome also entered into an Invention Assignment, Confidentiality, Non-Compete and Non-Solicit Agreement.
Beijing GenExosome is engaged in the development of exosome technology to improve diagnosis and management of diseases. Exosomes are tiny, subcellular, membrane-bound vesicles in diameter of 30-150 nm that are released by almost all cell types and that can carry membrane and cellular proteins, as well as genetic materials that are representative of the cell of origin. Profiling various bio-molecules in exosomes may serve as useful biomarkers for a wide variety of diseases. Beijing GenExosome’s research kits are designed to be used by researchers for biomarker discovery and clinical diagnostic development, and the advancement of targeted therapies. Currently, research kits and service are available to isolate exosomes or extract exosomal RNA/protein from serum/plasma, urine and saliva samples. Beijing GenExosome is seeking to decode proteomic and genomic alterations underlying a wide-range of pathologies, thus allowing for the introduction of novel non-invasive “liquid biopsies”. Its mission is focused toward diagnostic advancements in the fields of oncology, infectious diseases and fibrotic diseases, and discovery of disease-specific exosomes to provide disease origin insight necessary to enable personalized clinical management. There is no guarantee that Beijing GenExosome will be able to successfully achieve its stated mission.
On July 18, 2018, we formed a wholly owned subsidiary, Avactis Biosciences Inc. (“Avactis”), a Nevada corporation, which will be focused on accelerating commercial activities related to cellular therapies, including regenerative medicine with stem/progenitor cells as well as cellular immunotherapy including CAR-T, CAR-NK, TCR-T and others. The subsidiary is designed to integrate and optimize our global scientific and clinical resources to further advance the use of cellular therapies to treat certain cancers. On October 23, 2018, Avactis and Arbele Limited (“Arbele”) agreed to the establishment of AVAR BioTherapeutics (China) Co. Ltd. (“AVAR”), a Sino-foreign equity joint venture, pursuant to an Equity Joint Venture Agreement (the “AVAR Agreement”), which will be owned 60% by Avactis and 40% by Arbele. The purpose and business scope of the Joint Venture is to research, develop, produce, sell, distribute and generally commercialize CAR-T/CAR-NK/TCR-T/universal cellular immunotherapy in China. Avactis is required to contribute USD $10 million (or equivalent in RMB) in cash and/or services, which shall be contributed in tranches based on milestones to be determined jointly by AVAR and Avactis in writing subject to Avactis’ cash reserves. Within 30 days, Arbele shall make contribution of USD $6.66 million in the form of entering into a License Agreement with AVAR granting AVAR with an exclusive right and license in China to its technology and intellectual property pertaining to CAR-T/CAR-NK/TCR-T/universal cellular immunotherapy technology and any additional technology developed in the future with terms and conditions to be mutually agreed upon Avactis and AVAR and services. As of the date of this prospectus, AVAR is in process of being established and the License Agreement has not been finalized.
17
Issuer Purchases of Equity Securities
On March 27, 2018, we repurchased 520,000 shares of our common stock from a third party through a privately negotiated transaction at an aggregate price of $522,500, of which $2,500 was paid to an escrow agent as share repurchase cost.
Private Placement
From April 2018 through May 2018, we entered into subscription agreements with four accredited investors pursuant to which these investors purchased an aggregate of 3,107,000 shares of our common stock for a purchase price of $5,437,250. The closing occurred with respect to $3,500,000 on April 20, 2018, with respect to $157,500 on April 26, 2018, with respect to $997,500 on May 5, 2018 and with respect to $782,250 on May 24, 2018. In connection with this private placement, we are required to pay Boustead Securities, LLC, acting as placement agent, a cash fee of equal to 7% of the gross proceeds received by us from such closing and issue to the placement agent warrants to purchase common stock exercisable for a period of five years equal to 7% of the gross proceeds received by us from such closing, divisible by and exercisable at a strike price equal to 100% of the fair market value of our common stock as of the date of the closing. The warrants are not exercisable for more than five years from the effectiveness of the offering. Furthermore, the warrants may not be sold, transferred, assigned, pledged or hypothecated, or be the subject of any hedging, short sale, derivative, put or call transaction that would result in the effective economic disposition of the securities for a period of 180 days after the date of effectiveness or commencement of sales of the public offering, except as provided for in FINRA Rule 5110(g)(2). This restriction is imposed pursuant to the requirements of FINRA Rule 5110(g)(1). The warrant holder has a piggyback registration right on the warrant shares or the Company has obligation to include the resale of the warrant shares in its next registration statement other than those on Form S-8 or Form S-4; provided, the piggyback registration rights shall not last for more than seven years from the effective date of this registration statement pursuant to FINRA Rule 5110(f)(2)(G)(v).
DOING Biomedical Technology Co., Ltd. Investment
On April 23, 2018, we, Avalon Shanghai, Beijing DOING Biomedical Technology Co., Ltd., or DOING, and the accredited investor party to a subscription agreement with us executed on March 3, 2017 for a purchase price of $3,000,000, or the DOING Investment, entered into a Supplementary Agreement Related to Share Subscription pursuant to which Avalon Shanghai agreed to pay approximately USD $1,305,000 to DOING representing one-third of the DOING Investment plus 20% interest resulting in a reduction in the shares from the March 2017 transaction by one-third to 2,000,000 shares. On August 8, 2018, DOING the accredited investor party sold the remaining 2,000,000 shares of common stock to a third party in consideration of $2,000,000, thereby satisfying the repayment obligation in full.
Joint Venture - Epicon Biosciences Co., Ltd.
On May 29, 2018, Avalon Shanghai entered into a Joint Venture Agreement with Jiangsu Unicorn Biological Technology Co., Ltd., or Unicorn, pursuant to which a company named Epicon Biosciences Co., Ltd. (“Epicon”) was formed on August 14, 2018. Epicon is owned 60% by Unicorn and 40% by Avalon Shanghai. Within two years of execution of the Joint Venture Agreement, Unicorn shall invest cash into Epicon in an amount not less than RMB 8,000,000 (approximately $1.2 million) and the premises of the laboratories of Nanjing Hospital of Chinese Medicine for exclusive use by Epicon, and Avalon Shanghai shall invest cash into Epicon in an amount not less than RMB 10,000,000 (approximately $1.5 million). As of the date of this prospectus, Unicorn has invested the premises of the laboratories of Nanjing Hospital of Chinese Medicine and Avalon Shanghai has contributed RMB 3,000,000 (approximately $0.4 million). Epicon is focused on cell preparation, third party testing, biological sample repository for commercial and scientific research purposes and the clinical transformation of scientific achievements.
Avactis Biosciences
On July 18, 2018, we formed a wholly owned subsidiary, Avactis Biosciences, Inc., a Nevada corporation, which will be focused on accelerating commercial activities related to Chimeric Antigen Receptor (CAR)-T technologies. The new subsidiary is designed to integrate and optimize our global scientific and clinical resources to further advance the use of CAR-T to treat certain cancers.
Letter of Intent with Arbele Limited, a Hong Kong Company
On July 30, 2018, we signed a Letter of Intent with Arbele Limited, a Hong Kong company (“Arbele”) for a proposed strategic partnership agreement. The purpose of the proposed transaction is to form a joint venture company, AVAR BioTherapeutics (China) Co. Ltd., to develop, manufacture, and commercializing CAR-T immunotherapy for treating cancer patients in China, utilizing intellectual property from Arbele and the clinical platform of the LuDaopei Medical Group in China. We paid a $100,000 fee to Arbele for a five-month exclusive right to complete the definitive agreements for the transaction.
18
Strategic Partnership with Weill Cornell Medical College
On August 6, 2018, we entered into a strategic partnership agreement with Weill Cornell’s cGMP Cellular Therapy Facility and Laboratory for Advanced Cellular Engineering headed by Dr. Yen-Michael Hsu. This strategic partnership aims to co-develop bio-production and standardization procedures in procurement, storage, processing, clinical study protocols, and bio-banking for Chimeric Antigen Receptor (CAR)-T therapy, in accordance with the Foundation of Accreditation for Cellular Therapy (FACT) and American Association of Blood Banks (AABB) standards. This partnership also includes a CAR-T education program to support and foster collaborative research and training programs for scientists and clinicians between Weill Cornell and Hebei Yanda LuDaopei Hospital, which is our main affiliated clinical facility as well as the world’s single largest medical institution in CAR-T therapy.
Changes to Board of Directors
On June 4, 2018, Tevi Troy was appointed to the Board of Directors. Dr. Troy will receive options to acquire 40,000 shares of common stock per year commencing January 1, 2019 at an exercise price equal to the closing price on December 31st of the prior year. The options shall vest in equal amounts quarterly and shall be exercisable for a period of five years. For 2018, we granted Dr. Troy options to acquire 20,000 shares of common stock at an exercise price of $2.30 for a term of five years with 10,000 options vesting immediately and the balance vesting October 1, 2018. In addition, Dr. Troy will receive $5,000 per quarter for serving as chairman of the nominating and corporate governance committee commencing upon formation.
On July 5, 2018, William B. Stilley, III was appointed to the Board of Directors. Mr. Stilley will receive options to acquire 40,000 shares of common stock per year commencing January 1, 2019 at an exercise price equal to the closing price on December 31st of the prior year. The options shall vest in equal amounts quarterly and shall be exercisable for a period of five years. For 2018, we granted Mr. Stilley options to acquire 20,000 shares of common stock at an exercise price of $2.30 for a term of five years with 10,000 options vesting immediately and the balance vesting October 1, 2018. In addition, Mr. Stilley will receive $7,500 per quarter for serving as chairman of the audit committee commencing upon formation.
On July 9, 2018, Meng Li resigned as a director of the Company. Ms. Li will continue to serve as our Chief Operating Officer and Secretary and will also serve as an observer to the Board of Directors without voting capacity.
On July 30, 2018, Steven A. Sanders was appointed to the Board of Directors. Mr. Sanders will receive options to acquire 40,000 shares of common stock per year commencing January 1, 2019 at an exercise price equal to the closing price on December 31st of the prior year. The options shall vest in equal amounts quarterly and shall be exercisable for a period of five years. For 2018, we granted Mr. Sanders options to acquire 20,000 shares of common stock at an exercise price of $2.80 for a term of five years with 10,000 options vesting immediately and the balance vesting October 1, 2018. In addition, Mr. Sanders will receive $5,000 per quarter for serving as a member of our audit committee and nominating and corporate governance committee, respectively, commencing upon formation.
On July 30, 2018, Steven P. Sukel resigned as a director of the Company.
Public Offering
On August 13, 2018, we entered into an Underwriting Agreement with Boustead Securities, LLC. Pursuant to the Underwriting Agreement, on August 14, 2018, we closed a public offering in which it sold 939,450 shares of common stock at a per share price of $2.25 per share for total gross proceeds of $2,113,763 less commission of $105,689 resulting in net proceeds, before expenses, of $2,008,074.
19
Joint Venture – AVAR BioTherapeutics (China) Co. Ltd.
On October 23, 2018, Avactis Biosciences, Inc. (“Avactis”), a wholly-owned subsidiary of the Company, and Arbele Limited (“Arbele”) agreed to the establishment of AVAR BioTherapeutics (China) Co. Ltd. (“AVAR”), a Sino-foreign equity joint venture, pursuant to an Equity Joint Venture Agreement (the “AVAR Agreement”), which will be owned 60% by Avactis and 40% by Arbele. The purpose and business scope of the Joint Venture is to research, develop, produce, sell, distribute and generally commercialize CAR-T/CAR-NK/TCR-T/universal cellular immunotherapy in China. Avactis is required to contribute USD $10 million (or equivalent in RMB) in cash and/or services, which shall be contributed in tranches based on milestones to be determined jointly by AVAR and Avactis in writing subject to Avactis’ cash reserves. Within 30 days, Arbele shall make contribution of USD $6.66 million in the form of entering into a License Agreement with AVAR granting AVAR with an exclusive right and license in China to its technology and intellectual property pertaining to CAR-T/CAR-NK/TCR-T/universal cellular immunotherapy technology and any additional technology developed in the future with terms and conditions to be mutually agreed upon Avactis and AVAR and services.
In addition, Avactis is responsible for:
| ● | Contributing registered capital of RMB 5,000,000 (approximately $700,000) for working capital purposes as required by local regulation, which is not required to be contributed immediately and will be contributed subject to Avactis’ discretion; |
| ● | assist AVAR in setting up its business operations and obtaining all required permits and licenses from the Chinese government; |
| ● | assisting AVAR in recruiting, hiring and retaining personnel; |
| ● | providing AVAR with access to various hospital networks in China to assist in the testing and commercialization of the CAR-T/CAR-NK/TCR-T/universal cellular immunotherapy technology in China; |
| ● | assisting AVAR in managing the Good Manufacturing Practices (GMP) facility and clinic to be developed by AVAR; |
| ● | providing AVAR with advice pertaining to conducting clinicals in China; and |
| ● | Within 6 days of signing the AVAR Agreement, Avactis is required to pay to Arbele $300,000 as a research and development fee with an additional two payments of $300,000 (for a total of $900,000) to be paid upon mutually agreed upon milestones. |
Under AVAR Agreement, Arbele shall be responsible for the following:
| ● | Entering into a License Agreement with AVAR; and |
| ● | Providing AVAR with research and development expertise pertaining to clinical laboratory medicine when hired by AVAR. |
As of the date of this prospectus, Avactis has paid $300,000 to Arbele as research and development fee, AVAR is in process of being established and the License Agreement has not been finalized.
AVAR’s Board of Directors shall consist of three directors, of which two (2) directors shall be appointed by Avactis who shall initially be David Jin, M.D., Ph.D and one other director to be determined by Avactis and agreed to by Arbele. One director shall be appointed by Arbele who shall initially be John Luk, Dr. Med.Sc., EMBA.
Nasdaq Uplisting
On November 5, 2018, our common stock commenced trading on the NASDAQ Capital Market under the symbol “AVCO.”
20
An investment in our securities carries a significant degree of risk. You should carefully consider the following risks, as well as the other information contained in this prospectus, including our historical and pro forma financial statements and related notes included elsewhere in this prospectus, before you decide to purchase the securities. Any one of these risks and uncertainties has the potential to cause material adverse effects on our business, prospects, financial condition and operating results which could cause actual results to differ materially from any forward-looking statements expressed by us and a significant decrease in the value of our common stock. Refer to “Forward-Looking Statements”.
You should read the prospectus supplement and the documents incorporated herein by reference to see if there are additional risks that have arisen since the date of this prospectus or are specific to the terms of an offering
We may not be successful in preventing the material adverse effects that any of the following risks and uncertainties may cause. These potential risks and uncertainties may not be a complete list of the risks and uncertainties facing us. There may be additional risks and uncertainties that we are presently unaware of, or presently consider immaterial, that may become material in the future and have a material adverse effect on us. You could lose all or a significant portion of your investment due to any of these risks and uncertainties.
General Operating and Business Risks
Our limited operating history makes it difficult for us to evaluate our future business prospects and make decisions based on those estimates of our future performance.
We did not begin operations of our business through AHS until May 2015. We have a limited operating history and limited revenue. As a consequence, it is difficult, if not impossible, to forecast our future results based upon our historical data. Reliance on the historical results may not be representative of the results we will achieve, particularly in our combined form. Because of the uncertainties related to our lack of historical operations, we may be hindered in our ability to anticipate and timely adapt to increases or decreases in revenues or expenses. If we make poor budgetary decisions as a result of unreliable historical data, we could be less profitable or incur losses, which may result in a decline in our stock price.
Our results of operations have not resulted in profitability and we may not be able to achieve profitability going forward.
We incurred a net loss amounting to $4,049,645 for the year ended December 31, 2017 and a net loss amounting to $5,298,035 for the nine months ended September 30, 2018. If we incur additional significant losses, our stock price may decline, perhaps significantly. Our management is developing plans to achieve profitability. Our business plan is speculative and unproven. There is no assurance that we will be successful in executing our business plan or that even if we successfully implement our business plan, that we will be able to curtail our losses now or in the future. Further, as we are a new enterprise, we expect that net losses will continue.
We depend upon key personnel and need additional personnel.
Our success depends on the continuing services of Wenzhao Lu, our Chairman of the Board, and David Jin, Meng Li and Luisa Ingargiola, our executive officers. The loss of Mr. Lu, Dr. Jin, Ms. Li or Ms. Ingargiola could have a material and adverse effect on our business operations. Additionally, the success of our operations will largely depend upon our ability to successfully attract and maintain competent and qualified key management personnel. As with any company with limited resources, there can be no guaranty that we will be able to attract such individuals or that the presence of such individuals will necessarily translate into profitability for us. Our inability to attract and retain key personnel may materially and adversely affect our business operations.
Currently, we have a single consulting contract with a related party in China. The loss of such customer could adversely impact our financial condition and results of operations.
During the year ended December 31, 2017, we recognized an aggregate of $1,077,550 in revenue, of which $222,611 was generated from related parties. During the nine months ended September 30, 2018, we recognized an aggregate of $1,217,509 in revenue, of which $213,394 was generated from related parties. Wenzhao Lu, our Chairman and significant shareholder, is the Chairman of each of the related parties. Although we maintain close working relationships with our related parties, the consulting agreements with our related parties expired as of March 31, 2018. On April 1, 2018, Avalon Shanghai entered into an advisory service contract with Beijing Ludaopei Blood Disease Research Institute Co., Ltd., a subsidiary of the Daopei Hospital Group (a related party of ours). Under the terms of the contract, the aggregate amount of advisory service fees was $300,000, which was invoiced by the end of 2018. The contract expired on December 31, 2018. The loss of this related party customer, and our failure to replace such customer with other customers, could have a material adverse effect on our financial condition or results of operation.
21
Our auditors have issued a “going concern” audit opinion.
Our independent auditors have indicated, in their report on our December 31, 2017 consolidated financial statements, that there is substantial doubt about our ability to continue as a going concern. We had an accumulated deficit of $8,638,297 at September 30, 2018. We have a limited operating history and our continued growth is dependent upon the continuation of providing medical consulting services to our related parties, generating rental revenue from our income-producing real estate property in New Jersey and generating revenue from proprietary exosome isolation systems by developing proprietary diagnostic and therapeutic products leveraging exosome technology; hence generating revenues, and obtaining additional financing to fund future obligations and pay liabilities arising from normal business operations. Our ability to continue as a going concern is dependent on our ability to raise additional capital, implement our business plan, and generate significant revenues. There are no assurances that we will be successful in our efforts to generate significant revenues, maintain sufficient cash balance or report profitable operations or to continue as a going concern. We plan on raising capital through the sale of equity or debt instruments to implement our business plan. However, there is no assurance these plans will be realized and that any additional financings will be available to our company on satisfactory terms and conditions, if any.
We must effectively manage the growth of our operations, or our company will suffer.
To manage our growth, we believe we must continue to implement and improve our services and products. We may not have adequately evaluated the costs and risks associated with our planned expansion, and our systems, procedures, and controls may not be adequate to support our operations. In addition, our management may not be able to achieve the rapid execution necessary to successfully offer our products and services and implement our business plan on a profitable basis. The success of our future operating activities will also depend upon our ability to expand our support system to meet the demands of our growing business. Any failure by our management to effectively anticipate, implement, and manage changes required to sustain our growth would have a material adverse effect on our business, financial condition, and results of operations.
Our business requires substantial capital, and if we are unable to maintain adequate financing sources our profitability and financial condition will suffer and jeopardize our ability to continue operations.
In connection with the strategic development portion of our business, we will need significant capital in order to implement acquisitions of technologies. In addition, we will need a significant amount of capital in order to fully implement our advisory business, maintain our rental property and further develop our exosome business. If we are unable to maintain adequate financing or other sources of capital are not available, we could be forced to suspend, curtail or reduce our operations, which could harm our revenues, profitability, financial condition and business prospects.
Our revenue and results of operations may suffer if we are unable to attract new clients, continue to engage existing clients, or sell additional products and services.
We presently derive our revenue from providing medical related consulting services to a related party, generating rental revenue from our income-producing real estate property in New Jersey and generating revenue from proprietary exosome isolation systems by developing proprietary diagnostic and therapeutic products leveraging exosome technology. Our growth therefore depends on our ability to attract new clients, maintain existing clients and properties and sell additional products and services to existing clients. This depends on our ability to understand and anticipate market and pricing trends and our clients’ needs and our ability to deliver consistent, reliable, high-quality services. Our failure to engage new clients, continue to re-engage with our existing clients or cross-sell additional services could materially and adversely affect our operating results.
22
Our prospects will suffer if we are not able to hire, train, motivate, manage, and retain a significant number of highly skilled employees.
We only recently commenced business and we presently generate medical related consulting services to related parties, generating rental revenue from our income-producing real estate property in New Jersey and generating revenue from proprietary exosome isolation systems by developing proprietary diagnostic and therapeutic products leveraging exosome technology. On the consulting side, Wenzhao Lu, our Chairman and significant shareholder, is the Chairman of each of the clients in which we have provided consulting services. Our future success depends upon our ability to hire, train, motivate, manage, and retain a significant number of highly skilled employees, particularly research analysts, technical experts, and sales and marketing staff. We will experience competition for professional personnel in each of our business lines. Hiring, training, motivating, managing, and retaining employees with the skills we need is time consuming and expensive. Any failure by us to address our staffing needs in an effective manner could hinder our ability to continue to provide high-quality products and services and to grow our business.
Potential liability claims may adversely affect our business.
Our services, which may include recommendations and advice to organizations regarding complex business and operational processes and regulatory and compliance issues may give rise to liability claims by our clients or by third parties who bring claims against our clients. Healthcare organizations often are the subject of regulatory scrutiny and litigation, and we also may become the subject of such litigation based on our advice and services. Any such litigation, whether or not resulting in a judgment against us, may adversely affect our reputation and could have a material adverse effect on our financial condition and results of operations. We may not have adequate insurance coverage for claims against us.
In accordance with our strategic development policy, we may invest in companies for strategic reasons and may not realize a return on our investments.
Similar to the development of our majority-owned subsidiary, GenExosome, from time to time, we may make investments in companies. These investments may be for strategic objectives to support our key business initiatives but may also be standalone investments or acquisitions. Such investments or acquisitions could include equity or debt instruments in private companies, many of which may not be marketable at the time of our initial investment. These companies may range from early-stage companies that are often still defining their strategic direction to more mature companies with established revenue streams and business models. The success of these companies may depend on product development, market acceptance, operational efficiency, and other key business factors. The companies in which we invest may fail because they may not be able to secure additional funding, obtain favorable investment terms for future financings, or take advantage of liquidity events such as public offerings, mergers, and private sales. If any of these private companies fails, we could lose all or part of our investment in that company. If we determine that impairment indicators exist and that there are other-than-temporary declines in the fair value of the investments, we may be required to write down the investments to their fair value and recognize the related write-down as an investment loss.
Our growing operations in the PRC could expose us to risks that could have an adverse effect on our costs of operations.
Our client base is presently located in the PRC. We intend to grow this client base in the PRC as well as the United States. As a result, we expect to continue to add personnel in the PRC. With a significant focus of our operations in the PRC, our reliance on a workforce in the PRC exposes us to disruptions in the business, political, and economic environment in that region. Maintenance of a stable political environment between the PRC and the United States is important to our operations, and any disruption in this relationship may directly negatively affect our operations. Our operations in the PRC require us to comply with complex local laws and regulatory requirements and expose us to foreign currency exchange rate risk. Our operations may also be subject to reduced or inadequate protection of our intellectual property rights, and security breaches. Further, it may be difficult to transfer funds from our Chinese operations to our company. Negative developments in any of these areas could increase our costs of operations or otherwise harm our business.
23
We face intense competition which could cause us to lose market share.
In the healthcare markets in the United States and the People’s Republic of China, we will compete with large healthcare providers who have more significant financial resources, established market positions, long-standing relationships, and who have more significant name recognition, technical, marketing, sales, distribution, financial and other resources than we do. The resources available to our competitors to develop new services and products and introduce them into the marketplace exceed the resources currently available to us. This intense competitive environment may require us to make changes in our services, products, pricing, licensing, distribution, or marketing to develop a market position.
Our success is heavily dependent on protecting our intellectual property rights.
Through GenExosome, we own four patents in China with related trademarks. We are in the process of applying for those same patents and trademarks in the United States and are also in the process of developing additional patents and related intellectual property. We own and control a variety of trade secrets, confidential information, trademarks, trade names, copyrights, and other intellectual property rights that, in the aggregate, are of material importance to our business. We consider our trademarks, service marks, and other intellectual property to be proprietary, and rely on a combination of copyright, trademark, trade secret, non-disclosure, and contractual safeguards to protect our intellectual property rights. Our success will, in part, depend on our ability to obtain trademarks and patents. We have also entered into confidentiality agreements with our employees and consultants. We cannot be certain that others will not gain access to these trade secrets or that our patents will provide adequate protection. Others may independently develop substantially equivalent proprietary information and techniques or otherwise gain access to our trade secrets.
We may face uncertainty and difficulty in obtaining and enforcing our patents and other proprietary rights.
Our success will depend in large part on our ability to obtain, maintain, and defend patents on our product candidates, obtain licenses to use third-party technologies, protect our trade secrets and operate without infringing the proprietary rights of others. There can be no assurance that our pending patent applications will be approved, or that challenges will not be instituted against the validity or enforceability of any patent licensed-in or owned by us. Additionally, we have entered into various confidentiality agreements with employees and third parties. There is no assurance that such agreements will be honored by such parties or enforced in whole or part by the courts. The cost of litigation to uphold the validity and prevent infringement of a patent is substantial. Furthermore, there can be no assurance that others will not independently develop substantially equivalent technologies not covered by patents to which we have rights or obtain access to our know-how. In addition, the laws of certain countries may not adequately protect our intellectual property. Our competitors may possess or obtain patents on products or processes that are necessary or useful to the development, use, or manufacture of our product candidates. There can also be no assurance that our proposed technology will not infringe upon patents or proprietary rights owned by others, with the result that others may bring infringement claims against us and require us to license such proprietary rights, which may not be available on commercially reasonable terms, if at all. Any such litigation, if instituted, could have a material adverse effect, potentially including monetary penalties, diversion of management resources, and injunction against continued manufacture, use, or sale of certain products or processes.
We also rely upon non-patented proprietary know-how. There can be no assurance that we can adequately protect our rights in such non-patented proprietary know-how, or that others will not independently develop substantially equivalent proprietary information or techniques or gain access to our proprietary know-how. Any of the foregoing events could have a material adverse effect on us. In addition, if any of our trade secrets, know-how or other proprietary information were to be disclosed, or misappropriated, the value of our trade secrets, know-how and other proprietary rights would be significantly impaired and our business and competitive position would suffer.
In September 2011, the Leahy-Smith America Invents Act, or the Leahy-Smith Act, was signed into law. The Leahy-Smith Act includes a number of significant changes to U.S. patent law. These include provisions that affect the way patent applications will be prosecuted and may also affect patent litigation. In particular, under the Leahy-Smith Act, the United States transitioned in March 2013 to a “first to file” system in which the first inventor to file a patent application will be entitled to the patent. Third parties are allowed to submit prior art before the issuance of a patent by the U.S. Patent and Trademark Office, or USPTO, and may become involved in opposition, derivation, post-grant and inter partes review, or interference proceedings challenging our patent rights. An adverse determination in any such submission, proceeding or litigation could reduce the scope of, or invalidate, our patent rights, which could adversely affect our competitive position.
24
The USPTO has developed new and untested regulations and procedures to govern the full implementation of the Leahy-Smith Act, and many of the substantive changes to patent law associated with the Leahy-Smith Act, and in particular, the “first-to-file” provisions, only became effective in March 2013. The Leahy-Smith Act has also introduced procedures that may make it easier for third parties to challenge issued patents, as well as to intervene in the prosecution of patent applications. Finally, the Leahy-Smith Act contains new statutory provisions that still require the USPTO to issue new regulations for their implementation, and it may take the courts years to interpret the provisions of the new statute. Accordingly, it is not clear what, if any, impact the Leahy-Smith Act will have on the operation of our business. The Leahy-Smith Act and its implementation could increase the uncertainties and costs surrounding the prosecution of our patent applications and the enforcement or defense of our issued patents.
It is difficult and costly to protect our proprietary rights, and we may not be able to ensure their protection. If we fail to protect or enforce our intellectual property rights adequately or secure rights to patents of others, the value of our intellectual property rights would diminish.
Our commercial viability will depend in part on obtaining and maintaining patent protection and trade secret protection of our product candidates, and the methods used to manufacture them, as well as successfully defending these patents against third-party challenges. Our ability to stop third parties from making, using, selling, offering to sell, or importing our products is dependent upon the extent to which we have rights under valid and enforceable patents or trade secrets that cover these activities.
The patent positions of pharmaceutical and biopharmaceutical companies can be highly uncertain and involve complex legal and factual questions for which important legal principles remain unresolved. No consistent policy regarding the breadth of claims allowed in biopharmaceutical patents has emerged to date in the United States. The biopharmaceutical patent situation outside the United States is even more uncertain. Changes in either the patent laws or in interpretations of patent laws in the United States and other countries may diminish the value of our intellectual property. Accordingly, we cannot predict the breadth of claims that may be allowed or enforced in the patents we own. Further, if any of our patents are deemed invalid and unenforceable, it could impact our ability to commercialize or license our technology.
The degree of future protection for our proprietary rights is uncertain because legal means afford only limited protection and may not adequately protect our rights or permit us to gain or keep our competitive advantage. For example:
| ● | others may be able to make products that are similar to our product candidates but that are not covered by the claims of any of our patents; | |
| ● | we might not have been the first to make the inventions covered by any issued patents or patent applications we may have; | |
| ● | we might not have been the first to file patent applications for these inventions; | |
| ● | it is possible that any pending patent applications we may have will not result in issued patents; | |
| ● | any issued patents may not provide us with any competitive advantages, or may be held invalid or unenforceable as a result of legal challenges by third parties; | |
| ● | we may not develop additional proprietary technologies that are patentable or protectable under trade secrets law; or | |
| ● | the patents of others may have an adverse effect on our business. |
We also may rely on trade secrets to protect our technology, especially where we do not believe patent protection is appropriate or obtainable. However, trade secrets are difficult to protect. Although we use reasonable efforts to protect our trade secrets, our employees, consultants, contractors, outside scientific collaborators, and other advisors may unintentionally or willfully disclose our information to competitors. In addition, courts outside the United States are sometimes less willing to protect trade secrets. Moreover, our competitors may independently develop equivalent knowledge, methods, and know-how.
25
If any of our trade secrets, know-how or other proprietary information is disclosed, the value of our trade secrets, know-how and other proprietary rights would be significantly impaired and our business and competitive position would suffer.
Our viability also depends upon the skills, knowledge and experience of our scientific and technical personnel, and our consultants and advisors. To help protect our proprietary know-how and our inventions for which patents may be unobtainable or difficult to obtain, we rely on trade secret protection and confidentiality agreements. To this end, we require all of our employees, consultants, advisors and contractors to enter into agreements which prohibit unauthorized disclosure and use of confidential information and, where applicable, require disclosure and assignment to us of the ideas, developments, discoveries and inventions important to our business. These agreements are often limited in duration and may not provide adequate protection for our trade secrets, know-how or other proprietary information in the event of any unauthorized use or disclosure or the lawful development by others of such information. In addition, enforcing a claim that a third party illegally obtained and is using any of our trade secrets is expensive and time consuming, and the outcome is unpredictable. If any of our trade secrets, know-how or other proprietary information is improperly disclosed, the value of our trade secrets, know-how and other proprietary rights would be significantly impaired and our business and competitive position would suffer.
We may incur substantial costs as a result of litigation or other proceedings relating to patent and other intellectual property rights and we may be unable to protect our rights to, or use of, our technology.
If we choose to go to court to stop a third party from using the inventions claimed in our patents, that individual or company has the right to ask the court to rule that such patents are invalid and/or should not be enforced against that third party. These lawsuits are expensive and would consume time and other resources, even if we were successful in discontinuing the infringement of our patents. In addition, there is a risk that the court will decide that these patents are not valid and that we do not have the right to stop the other party from using the inventions. There is also the risk that, even if the validity of these patents is upheld, the court will refuse to stop the other party on the ground that such other party’s activities do not infringe our rights to these patents. In addition, the U.S. Supreme Court has in the past invalidated tests used by the USPTO in granting patents over the past 20 years. As a consequence, issued patents may be found to contain invalid claims according to the newly revised standards. Some of our own patents may be subject to challenge and subsequent invalidation in a variety of post-grant proceedings, particularly inter partes review, before the USPTO or during litigation under the revised criteria, which make it more difficult to defend the validity of claims in already issued patents.
Furthermore, a third party may claim that we or our manufacturing or commercialization partners are using inventions covered by the third party’s patent rights and may go to court to stop us from engaging in our normal operations and activities, including making or selling our product candidates. These lawsuits are costly and could affect our results of operations and divert the attention of managerial and technical personnel. There is a risk that a court could decide that we or our commercialization partners are infringing the third party’s patents and order us or our partners to stop the activities covered by the patents. In addition, there is a risk that a court could order us or our partners to pay the other party damages for having violated the other party’s patents. The biotechnology industry has produced a proliferation of patents, and it is not always clear to industry participants, including us, which patents cover various types of products, manufacturing processes or methods of use. The coverage of patents is subject to interpretation by the courts, and the interpretation is not always uniform. If we are sued for patent infringement, we would need to demonstrate that our products, manufacturing processes or methods of use either do not infringe the patent claims of the relevant patent and/or that the patent claims are invalid, and we may not be able to do this. Proving invalidity, in particular, is difficult since it requires a showing of clear and convincing evidence to overcome the presumption of validity enjoyed by issued patents.
As some patent applications in the United States may be maintained in secrecy until the patents are issued, because patent applications in the United States and many foreign jurisdictions are typically not published until eighteen months after filing, and because publications in the scientific literature often lag behind actual discoveries, we cannot be certain that others have not filed patent applications for technology covered by our issued patents or our pending applications, or that we were the first to invent the technology. Our competitors may have filed, and may in the future file, patent applications covering technology similar to ours. Any such patent applications may have priority over our patent applications or patents, which could further require us to obtain rights to issued patents covering such technologies. If another party has filed a United States patent application on inventions similar to ours, we may have to participate in an interference proceeding declared by the USPTO to determine priority of invention in the United States. The costs of these proceedings could be substantial, and it is possible that such efforts would be unsuccessful if, unbeknownst to us, the other party had independently arrived at the same or similar invention prior to our own invention, resulting in a loss of our U.S. patent position with respect to such inventions.
26
Some of our competitors may be able to sustain the costs of complex patent litigation more effectively than we can because they have substantially greater resources. In addition, any uncertainties resulting from the initiation and continuation of any litigation or inter partes review proceedings could have a material adverse effect on our ability to raise the funds necessary to continue our operations.
Some jurisdictions in which we operate have enacted legislation which allows members of the public to access information under statutes similar to the U.S. Freedom of Information Act. Even though we believe our information would be excluded from the scope of such statutes, there are no assurances that we can protect our confidential information from being disclosed under the provisions of such laws. If any confidential or proprietary information is released to the public, such disclosures may negatively impact our ability to protect our intellectual property rights.
Breaches or compromises of our information security systems or our information technology systems or infrastructure could result in exposure of private information, disruption of our business and damage to our reputation, which could harm our business, results of operation and financial condition.
We utilize information security and information technology systems and websites that allow for the secure storage and transmission of proprietary or private information regarding our clients, patients, employees, vendors and others, including individually identifiable health information. A security breach of our network, hosted service providers, or vendor systems, may expose us to a risk of loss or misuse of this information, litigation and potential liability. Hackers and data thieves are increasingly sophisticated and operate large-scale and complex automated attacks, including on companies within the healthcare industry. Although we believe that we take appropriate measures to safeguard sensitive information within our possession, we may not have the resources or technical sophistication to anticipate or prevent rapidly-evolving types of cyber-attacks targeted at us, our clients, our patients, or others who have entrusted us with information. Actual or anticipated attacks may cause us to incur costs, including costs to deploy additional personnel and protection technologies, train employees, and engage third-party experts and consultants. We invest in industry standard security technology to protect personal information. Advances in computer capabilities, new technological discoveries, or other developments may result in the technology used by us to protect personal information or other data being breached or compromised. To our knowledge, we have not experienced any material breach of our cybersecurity systems. If our or our third-party service provider systems fail to operate effectively or are damaged, destroyed, or shut down, or there are problems with transitioning to upgraded or replacement systems, or there are security breaches in these systems, any of the aforementioned could occur as a result of natural disasters, software or equipment failures, telecommunications failures, loss or theft of equipment, acts of terrorism, circumvention of security systems, or other cyber-attacks, we could experience delays or decreases in revenue, and reduced efficiency of our operations. Additionally, any of these events could lead to violations of privacy laws, loss of customers, or loss, misappropriation or corruption of confidential information, trade secrets or data, which could expose us to potential litigation, regulatory actions, sanctions or other statutory penalties, any or all of which could adversely affect our business, and cause us to incur significant losses and remediation costs.
We may be exposed to liabilities under the Foreign Corrupt Practices Act, and any determination that we violated the Foreign Corrupt Practices Act or Chinese anti-corruption law could have a material adverse effect on our business.
We are subject to the Foreign Corrupt Practice Act, or FCPA, and other laws that prohibit improper payments or offers of payments to foreign governments and their officials and political parties by U.S. persons and issuers as defined by the statute for the purpose of obtaining or retaining business. Chinese anti-corruption law also strictly prohibits bribery of government officials. We have operations, agreements with third parties and make sales in China, where corruption may occur. Our activities in China create the risk of unauthorized payments or offers of payments by one of the employees, consultants, sales agents or distributors of our company, even though these parties are not always subject to our control. It is our policy to implement safeguards to prevent these practices by our employees. However, our existing safeguards and any future improvements may prove to be less than effective, and the employees, consultants, sales agents or distributors of our company may engage in conduct for which we might be held responsible.
27
Violations of the FCPA or other anti-corruption laws may result in severe criminal or civil sanctions, and we may be subject to other liabilities, which could negatively affect our business, operating results and financial condition. In addition, the United States government may seek to hold our company liable for successor liability FCPA violations committed by companies in which we invest or that we acquire.
Risk Factors Related to Clinical and Commercialization Activity
Our product candidates will require substantial time and resources in order to be developed, and there is no guarantee that we will develop them successfully.
Our exosome isolation system is in the early stage of production and use. The therapeutic products that we plan to develop as a byproduct of our isolation system will require substantial additional research and development time and expense, and certain products may require extensive clinical trials and perhaps additional pre-clinical testing, prior to commercialization, which may never occur. There can be no assurance that product candidates will be developed successfully, perform in the manner anticipated, or be commercially viable.
We may not be able to file INDs to commence additional clinical trials on the timelines we expect, and even if we are able to do so, the FDA may not permit us to proceed.
We hope to file a number of investigational new drug applications, or INDs, for cell based therapies and diagnostic systems through INDs over the next several years. However, the timing of our filing of these INDs is primarily dependent on receiving further data from our pre-clinical studies, and our timing of filing on all product candidates is subject to further research. Additionally, our submission of INDs is contingent upon having sufficient financial resources to prepare and complete the application.
We cannot be sure that submission of an IND will result in the United States Food and Drug Administration, or FDA, allowing further clinical trials to begin, or that, once begun, issues will not arise that result in the suspension or termination of such clinical trials. Any IND we submit could be denied by the FDA or the FDA could place any future investigation of ours on clinical hold until we provide additional information, either before or after clinical trials are initiated. Additionally, even if such regulatory authorities agree with the design and implementation of the clinical trials set forth in an IND or clinical trial application, we cannot guarantee that such regulatory authorities will not change their requirements in the future. Unfavorable future trial results or other factors, such as insufficient capital to continue development of a product candidate or program, could also cause us to voluntarily withdraw an effective IND.
We have limited experience in conducting clinical trials.
We have limited human clinical trial experience with respect to our product candidates. Although our CEO, Dr. David Jin, is formerly with the FDA, this will not provide assurance of success. The clinical testing process is governed by stringent regulation and is highly complex, costly, time-consuming, and uncertain as to outcome, and pharmaceutical products and products used in the regeneration of tissue may invite particularly close scrutiny and requirements from the FDA and other regulatory bodies. Our failure or the failure of our collaborators to conduct human clinical trials successfully or our failure to capitalize on the results of human clinical trials for our product candidates would have a material adverse effect on us. If our clinical trials of our product candidates or future product candidates do not sufficiently enroll or produce results necessary to support regulatory approval in the United States or elsewhere, or if they show undesirable side effects, we will be unable to commercialize these product candidates.
To receive regulatory approval for the commercial sale of our product candidates, we must conduct adequate and well-controlled clinical trials to demonstrate efficacy and safety in humans. Clinical failure can occur at any stage of the testing. Our clinical trials may produce negative or inconclusive results, and we may decide, or regulators may require us, to conduct additional clinical and/or non-clinical testing. In addition, the results of our clinical trials may show that our product candidates are ineffective or may cause undesirable side effects, which could interrupt, delay or halt clinical trials, resulting in the denial of regulatory approval by the FDA and other regulatory authorities. In addition, negative, delayed or inconclusive results may result in:
| ● | the withdrawal of clinical trial participants; |
| ● | the termination of clinical trial sites or entire trial programs; |
28
| ● | costs of related litigation; |
| ● | substantial monetary awards to patients or other claimants; |
| ● | impairment of our business reputation; |
| ● | loss of revenues; and |
| ● | the inability to commercialize our product candidates. |
Delays in the commencement, enrollment, and completion of clinical testing could result in increased costs to us and delay or limit our ability to obtain regulatory approval for our product candidates.
Delays in the commencement, enrollment or completion of clinical testing could significantly affect our product development costs. A clinical trial may be suspended or terminated by us, the FDA, or other regulatory authorities due to a number of factors. The commencement and completion of clinical trials require us to identify and maintain a sufficient number of trial sites, many of which may already be engaged in other clinical trial programs for the same indication as our product candidates. We may be required to withdraw from a clinical trial as a result of changing standards of care, or we may become ineligible to participate in clinical studies. We do not know whether planned clinical trials will begin on time or be completed on schedule, if at all. The commencement, enrollment and completion of clinical trials can be delayed for a number of reasons, including, but not limited to, delays related to:
| ● | findings in pre-clinical studies; |
| ● | reaching agreements on acceptable terms with prospective clinical research organizations, or CROs, and trial sites, the terms of which can be subject to extensive negotiation and may vary significantly among different CROs and trial sites; |
| ● | obtaining regulatory approval to commence a clinical trial; |
| ● | complying with conditions imposed by a regulatory authority regarding the scope or term of a clinical trial, or being required to conduct additional trials before moving on to the next phase of trials; |
| ● | obtaining institutional review board, or IRB, approval to conduct a clinical trial at numerous prospective sites; |
| ● | recruiting and enrolling patients to participate in clinical trials for a variety of reasons, including the size of the patient population, nature of trial protocol, meeting the enrollment criteria for our studies, screening failures, the inability of the sites to conduct trial procedures properly, the availability of approved effective treatments for the relevant disease and competition from other clinical trial programs for similar indications; |
| ● | retaining patients who have initiated their participation in a clinical trial but may be prone to withdraw due to the treatment protocol, lack of efficacy, personal issues, or side effects from the therapy, or who are lost to further follow-up; |
29
| ● | manufacturing sufficient quantities of a product candidate for use in clinical trials on a timely basis; |
| ● | complying with design protocols of any applicable special protocol assessment we receive from the FDA; |
| ● | severe or unexpected cell therapy side effects experienced by patients in a clinical trial; |
| ● | collecting, analyzing and reporting final data from the clinical trials; |
| ● | breaches in quality of manufacturing runs that compromise all or some of the doses made; positive results in FDA-required viral testing; karyotypic abnormalities in our cell product; or contamination in our manufacturing facilities, all of which events would necessitate disposal of all cells made from that source; |
| ● | availability of materials provided by third parties necessary to manufacture our product candidates; |
| ● | availability of adequate amounts of acceptable tissue for preparation of master cell banks for our products; and |
| ● | requirements to conduct additional trials and studies, and increased expenses associated with the services of our CROs and other third parties. |
If we are required to conduct additional clinical trials or other testing of our product candidates beyond those that we currently contemplate, we or our development partners, if any, may be delayed in obtaining, or may not be able to obtain or maintain, clinical or marketing approval for these product candidates. We may not be able to obtain approval for indications that are as broad as intended, or we may be able to obtain approval only for indications that are entirely different from those indications for which we sought approval.
Changes in regulatory requirements and guidance may occur, and we may need to amend clinical trial protocols to reflect these changes with appropriate regulatory authorities. Amendments may require us to resubmit our clinical trial protocols to IRBs for re-examination, which may impact the costs, timing, or successful completion of a clinical trial. If we experience delays in the completion of, or if we terminate, our clinical trials, the commercial prospects for our product candidates will be harmed, and our ability to generate product revenues will be delayed. In addition, many of the factors that cause, or lead to, a delay in the commencement or completion of clinical trials may also ultimately lead to the denial of regulatory approval of a product candidate. Even if we are able to ultimately commercialize our product candidates, other therapies for the same or similar indications may have been introduced to the market and already established a competitive advantage. Any delays in obtaining regulatory approvals may:
| ● | delay commercialization of, and our ability to derive product revenues from, our product candidates; |
| ● | impose costly procedures on us; or |
| ● | diminish any competitive advantages that we may otherwise enjoy. |
30
Our success depends upon the viability of our product candidates and we cannot be certain any of them will receive regulatory approval to be commercialized.
We will need FDA approval to market and sell any of our product candidates in the United States and approvals from FDA-equivalent regulatory authorities in foreign jurisdictions to commercialize our product candidates in those jurisdictions. In order to obtain FDA approval of any of our product candidates, we must submit to the FDA a new drug application, or NDA, or a biologics license application, or BLA, demonstrating that the product candidate is safe for humans and effective for its intended use. This demonstration requires significant research and animal tests, which are referred to as pre-clinical studies, as well as human tests, which are referred to as clinical trials. Satisfaction of the FDA’s regulatory requirements typically takes many years, depends upon the type, complexity, and novelty of the product candidate, and requires substantial resources for research, development, testing and manufacturing. We cannot predict whether our research and clinical approaches will result in cell therapies that the FDA considers safe for humans and effective for indicated uses. The FDA has substantial discretion in the drug approval process and may require us to conduct additional pre-clinical and clinical testing or to perform post-marketing studies. The approval process may also be delayed by changes in government regulation, future legislation, administrative action or changes in FDA policy that occur prior to or during our regulatory review.
Even if we comply with all FDA requests, the FDA may ultimately reject one or more of our NDAs or BLAs, as applicable. We cannot be sure that we will ever obtain regulatory clearance for our product candidates. Failure to obtain FDA approval of any of our product candidates will reduce our number of potentially salable products and, therefore, corresponding product revenues, and will have a material and adverse impact on our business.
As the results of earlier pre-clinical studies or clinical trials are not necessarily predictive of future results, any product candidate we advance into clinical trials may not have favorable results in later clinical trials or receive regulatory approval.
Even if our pre-clinical studies and clinical trials are completed as planned, clinical trials, we cannot be certain that their results will support the claims of our product candidates. Positive results in pre-clinical testing and early clinical trials do not ensure that results from later clinical trials will also be positive, and we cannot be sure that the results of later clinical trials will replicate the results of prior clinical trials and pre-clinical testing. A number of companies in the pharmaceutical industry, including those with greater resources and experience, have suffered significant setbacks in Phase II or Phase III clinical trials, even after seeing promising results in earlier clinical trials.
Our clinical trial process may fail to demonstrate that our product candidates are safe for humans and effective for indicated uses. This failure would cause us to abandon a product candidate and may delay development of other product candidates. Any delay in, or termination of, our clinical trials will delay or cause us to refrain from the filing of our NDAs and/or BLAs with the FDA and, ultimately, our ability to commercialize our product candidates and generate product revenues. In addition, our clinical trials to date involve small patient populations. Because of the small sample size, the results of these clinical trials may not be indicative of future results.
Our business faces significant government regulation, and there is no guarantee that our product candidates will receive regulatory approval.
Our research and development activities, pre-clinical studies, anticipated human clinical trials, and anticipated manufacturing and marketing of our potential products are subject to extensive regulation by the FDA and other regulatory authorities in the United States, as well as by regulatory authorities in other countries. In the United States, our product candidates are subject to regulation as biological products or as combination biological products/medical devices under the Federal Food, Drug and Cosmetic Act, the Public Health Service Act and other statutes, as outlined in the Code of Federal Regulations. Different regulatory requirements may apply to our products depending on how they are categorized by the FDA under these laws. These regulations can be subject to substantial and significant interpretation, addition, amendment or revision by the FDA and by the legislative process. The FDA may determine that we will need to undertake clinical trials beyond those currently planned. Furthermore, the FDA may determine that results of clinical trials do not support approval for the product. Similar determinations may be encountered in foreign countries. The FDA will continue to monitor products in the market after approval, if any, and may determine to withdraw its approval or otherwise seriously affect the marketing efforts for any such product. The same possibilities exist for trials to be conducted outside of the United States that are subject to regulations established by local authorities and local law. Any such determinations would delay or deny the introduction of our product candidates to the market and have a material adverse effect on our business, financial condition, and results of operations.
31
Cell based therapeutics are subject to ongoing periodic unannounced inspection by the FDA, the Drug Enforcement Agency, other federal agencies and corresponding state agencies to ensure strict compliance with good manufacturing practices, and other government regulations and corresponding foreign standards. We do not have control over third-party manufacturers’ compliance with these regulations and standards, nor can we guarantee that we will maintain compliance with such regulations in regards to our own manufacturing processes. Other risks include:
| ● | regulatory authorities may require the addition of labeling statements, specific warnings, a contraindication, or field alerts to physicians and pharmacies; |
| ● | regulatory authorities may withdraw their approval of the IND or the product or require us to take our approved products off the market; |
| ● | we may be required to change the way the product is manufactured or administered and we may be required to conduct additional clinical trials or change the labeling of our products; |
| ● | we may have limitations on how we promote our products; and |
| ● | we may be subject to litigation or product liability claims. |
Even if our product candidates receive regulatory approval in the United States, we may never receive approval or commercialize our product candidates outside of the United States. In order to market and commercialize any product candidate outside of the United States, we must establish and comply with numerous and varying regulatory requirements of other countries regarding manufacturing, safety and efficacy. Approval procedures vary among countries and can involve additional product testing and additional administrative review periods. The time required to obtain approval in other countries might differ from that required to obtain FDA approval. The regulatory approval process in other countries may include all of the risks detailed above regarding FDA approval in the United States as well as other risks. Regulatory approval in one country does not ensure regulatory approval in another, but a failure or delay in obtaining regulatory approval in one country may have a negative effect on the regulatory approval process in others. Failure to obtain regulatory approval in other countries, or any delay or setback in obtaining such approval, could have the same adverse effects detailed above regarding FDA approval in the United States. Such effects include the risks that our product candidates may not be approved for all indications requested, which could limit the uses of our product candidates and have an adverse effect on product sales and potential royalties, and that such approval may be subject to limitations on the indicated uses for which the product may be marketed or require costly, post-marketing follow-up studies.
Even if our product candidates receive regulatory approval, we may still face future development and regulatory difficulties.
Even if U.S. regulatory approval is obtained, the FDA may still impose significant restrictions on a product’s indicated uses or marketing, or impose ongoing requirements for potentially costly post-approval studies. If any of our products were granted accelerated approval, FDA could require post-marketing confirmatory trials to verify and describe the anticipated effect on irreversible morbidity or mortality or other clinical benefit. FDA may withdraw approval of a drug or indication approved under the accelerated approval pathway if a trial required to verify the predicted clinical benefit of the product fails to verify such benefit; other evidence demonstrates that the product is not shown to be safe or effective under the conditions of use; the applicant fails to conduct any required post-approval trial of the drug with due diligence; or the applicant disseminates false or misleading promotional materials relating to the product. In addition, the FDA currently requires as a condition for accelerated approval the pre-approval of promotional materials, which could adversely impact the timing of the commercial launch of the product.
Given the number of recent high-profile adverse safety events with certain drug and cell related products, the FDA may require, as a condition of approval, costly risk management programs, which may include safety surveillance, restricted distribution and use, patient education, enhanced labeling, special packaging or labeling, expedited reporting of certain adverse events, pre-approval of promotional materials, and restrictions on direct-to-consumer advertising. Furthermore, heightened Congressional scrutiny on the adequacy of the FDA’s drug approval process and the FDA’s efforts to assure the safety of marketed cell based therapy has resulted in the proposal of new legislation addressing drug safety issues. If enacted, any new legislation could result in delays or increased costs during the period of product development, clinical trials, and regulatory review and approval, as well as increased costs to assure compliance with any new post-approval regulatory requirements. Any of these restrictions or requirements could force us to conduct costly studies or increase the time for us to become profitable. For example, any labeling approved for any of our product candidates may include a restriction on the term of its use, or it may not include one or more of our intended indications.
32
Our product candidates will also be subject to ongoing FDA requirements for the labeling, packaging, storage, advertising, promotion, record-keeping, and submission of safety and other post-market information on the cell based therapy. New issues may arise during a product lifecycle that did not exist, or were unknown, at the time of product approval, such as adverse events of unanticipated severity or frequency, or problems with the facility where the product is manufactured. Since approved products, manufacturers, and manufacturers’ facilities are subject to continuous review and periodic inspections, these new issues post-approval may result in voluntary actions by us or may result in a regulatory agency imposing restrictions on that product or us, including requiring withdrawal of the product from the market or for use in a clinical study. If our product candidates fail to comply with applicable regulatory requirements, such as good manufacturing practices, a regulatory agency may:
| ● | issue warning letters; |
| ● | require us to enter into a consent decree, which can include imposition of various fines, reimbursements for inspection costs, required due dates for specific actions, and penalties for noncompliance; |
| ● | impose other civil or criminal penalties; |
| ● | suspend regulatory approval; |
| ● | suspend any ongoing clinical trials; |
| ● | refuse to approve pending applications or supplements to approved applications filed by us; |
| ● | impose restrictions on operations, including costly new manufacturing requirements; or |
| ● | seize or detain products or require a product recall. |
If we or current or future collaborators, manufacturers, or service providers fail to comply with healthcare laws and regulations, we or they could be subject to enforcement actions and substantial penalties, which could affect our ability to develop, market and sell our products and may harm our reputation.
Although we do not currently have any products on the market, once our therapeutic candidates or clinical trials are covered by federal health care programs, we will be subject to additional healthcare statutory and regulatory requirements and enforcement by the federal, state and foreign governments of the jurisdictions in which we conduct our business. Healthcare providers, physicians and third party payors play a primary role in the recommendation and prescription of any therapeutic candidates for which we obtain marketing approval. Our future arrangements with third party payors and customers may expose us to broadly applicable fraud and abuse, transparency, and other healthcare laws and regulations that may constrain the business or financial arrangements and relationships through which we market, sell and distribute our therapeutic candidates for which we obtain marketing approval. Restrictions under applicable federal and state healthcare laws and regulations include, but are not limited to, the following:
| ● | the U.S. federal Anti-Kickback Statute, which prohibits, among other things, persons from soliciting, receiving, offering or providing remuneration, directly or indirectly, to induce either the referral of an individual for a healthcare item or service, or the purchasing or ordering of an item or service, for which payment may be made, in whole or in part, under a federal healthcare program such as Medicare or Medicaid; |
33
| ● | federal civil and criminal false claims laws and civil monetary penalty laws, such as the U.S. federal FCA, which imposes criminal and civil penalties, including through civil whistleblower or qui tam actions, against, individuals or entities for knowingly presenting or causing to be presented, to the federal government, claims for payment that are false or fraudulent or making a false statement to avoid, decrease or conceal an obligation to pay money to the federal government. In addition, the government may assert that a claim including items and services resulting from a violation of the federal Anti-Kickback Statute constitutes a false or fraudulent claim for purposes of the FCA; |
| ● | HIPAA includes a fraud and abuse provision referred to as the HIPAA All-Payor Fraud Law, which imposes criminal and civil liability for executing a scheme to defraud any healthcare benefit program, or knowingly and willfully falsifying, concealing or covering up a material fact or making any materially false statement in connection with the delivery of or payment for healthcare benefits, items or services. Similar to the federal Anti-Kickback Statute, a person or entity does not need to have actual knowledge of the statute or specific intent to violate it in order to have committed a violation; |
| ● | HIPAA, as amended by HITECH, and its implementing regulations, which impose obligations on certain covered entity healthcare providers, health plans, and healthcare clearinghouses as well as their business associates that perform certain services involving the use or disclosure of individually identifiable health information, including mandatory contractual terms, with respect to safeguarding, the privacy, security, and transmission of individually identifiable health information, and require notification to affected individuals and regulatory authorities of certain breaches of security of individually identifiable health information; |
| ● | federal and state consumer protection and unfair competition laws, which broadly regulate marketplace activities and activities that potentially harm consumers; |
| ● | the federal Physician Payment Sunshine Act and the implementing regulations, also referred to as “Open Payments,” issued under the ACA, which require that manufacturers of pharmaceutical and biological drugs reimbursable under Medicare, Medicaid, and Children’s Health Insurance Programs report to the Department of Health and Human Services all consulting fees, travel reimbursements, research grants, and other payments, transfers of value or gifts made to physicians and teaching hospitals with limited exceptions; and |
| ● | analogous state laws and regulations, such as, state anti-kickback and false claims laws potentially applicable to sales or marketing arrangements and claims involving healthcare items or services reimbursed by nongovernmental third party payors, including private insurers; and some state laws require pharmaceutical companies to comply with the pharmaceutical industry’s voluntary compliance guidelines and the relevant compliance guidance promulgated by the federal government in addition to requiring drug and cell based therapy manufacturers to report information related to payments to physicians and other healthcare providers or marketing expenditures, and state laws governing the privacy and security of health information in certain circumstances, many of which differ from each other in significant ways and often are not preempted by HIPAA, thus complicating compliance efforts. |
The scope and enforcement of each of these laws is uncertain and subject to rapid change in the current environment of healthcare reform, especially in light of the lack of applicable precedent and regulations. Federal and state enforcement bodies have recently increased their scrutiny of interactions between healthcare companies and healthcare providers, which has led to a number of investigations, prosecutions, convictions and settlements in the healthcare industry. Responding to investigations can be time-and resource-consuming and can divert management’s attention from the business. Any such investigation or settlement could increase our costs or otherwise have an adverse effect on our business.
34
Ensuring that our business arrangements with third-parties comply with applicable healthcare laws and regulations could involve substantial costs. If our operations are found to be in violation of any such requirements, we may be subject to penalties, including civil or criminal penalties, monetary damages, the curtailment or restructuring of our operations, or exclusion from participation in government contracting, healthcare reimbursement or other government programs, including Medicare and Medicaid, any of which could adversely affect our financial results. Although effective compliance programs can mitigate the risk of investigation and prosecution for violations of these laws, these risks cannot be entirely eliminated. Any action against us for an alleged or suspected violation could cause us to incur significant legal expenses and could divert our management’s attention from the operation of our business, even if our defense is successful. In addition, achieving and sustaining compliance with applicable laws and regulations may be costly to us in terms of money, time and resources.
Any cell based therapies we develop may become subject to unfavorable pricing regulations, third party coverage and reimbursement practices or healthcare reform initiatives, thereby harming our business.
The regulations that govern marketing approvals, pricing, coverage and reimbursement for new drugs and cell based therapies vary widely from country to country. Some countries require approval of the sale price of a drug before it can be marketed. In many countries, the pricing review period begins after marketing or product licensing approval is granted. In some foreign markets, prescription pharmaceutical pricing remains subject to continuing governmental control even after initial approval is granted. Although we intend to monitor these regulations, our programs are currently in earlier stages of development and we will not be able to assess the impact of price regulations for a number of years. As a result, we might obtain regulatory approval for a product in a particular country, but then be subject to price regulations that delay our commercial launch of the product and negatively impact the revenues we are able to generate from the sale of the product in that country.
Our ability to commercialize any products successfully also will depend in part on the extent to which coverage and reimbursement for these products and related treatments will be available from government health administration authorities, private health insurers and other organizations. However, there may be significant delays in obtaining coverage for newly-approved cell based therapies. Moreover, eligibility for coverage does not necessarily signify that a cell based therapy will be reimbursed in all cases or at a rate that covers our costs, including research, development, manufacture, sale and distribution costs. Also, interim payments for new cell based therapy if applicable, may be insufficient to cover our costs and may not be made permanent. Thus, even if we succeed in bringing one or more products to the market, these products may not be considered medically necessary or cost-effective, and the amount reimbursed for any products may be insufficient to allow us to sell our products on a competitive basis. Because our programs are in earlier stages of development, we are unable at this time to determine their cost effectiveness, or the likely level or method of reimbursement. In addition, obtaining coverage and reimbursement approval of a product from a government or other third-party payor is a time-consuming and costly process that could require us to provide to each payor supporting scientific, clinical and cost-effectiveness data for the use of our product on a payor-by-payor basis, with no assurance that coverage and adequate reimbursement will be obtained. A payor’s decision to provide coverage for a product does not imply that an adequate reimbursement rate will be approved. Further, one payor’s determination to provide coverage for a product does not assure that other payors will also provide coverage for the product. Adequate third-party reimbursement may not be available to enable us to maintain price levels sufficient to realize an appropriate return on our investment in product development. If reimbursement is not available or is available only at limited levels, we may not be able to successfully commercialize any product candidate that we successfully develop.
Increasingly, the third party payors who reimburse patients or healthcare providers, such as government and private insurance plans, are seeking greater upfront discounts, additional rebates and other concessions to reduce the prices for pharmaceutical products. If the price we are able to charge for any products we develop, or the reimbursement provided for such products, is inadequate in light of our development and other costs, our return on investment could be adversely affected.
We currently expect that certain drugs we develop may need to be administered under the supervision of a physician on an outpatient basis. Under currently applicable U.S. law, certain drugs that are not usually self-administered (including injectable cell based therapies) may be eligible for coverage under Medicare through Medicare Part B. Specifically, Medicare Part B coverage may be available for eligible beneficiaries when the following, among other requirements have been satisfied:
| ● | the product is reasonable and necessary for the diagnosis or treatment of the illness or injury for which the product is administered according to accepted standards of medical practice; |
35
| ● | the product is typically furnished incident to a physician’s services; |
| ● | the indication for which the product will be used is included or approved for inclusion in certain Medicare-designated pharmaceutical compendia (when used for an off-label use); and |
| ● | the product has been approved by the FDA. |
Average prices for cell therapies may be reduced by mandatory discounts or rebates required by government healthcare programs or private payors and by any future relaxation of laws that presently restrict imports of drugs and cell based therapy from countries where they may be sold at lower prices than in the U.S. Reimbursement rates under Medicare Part B would depend in part on whether the newly approved product would be eligible for a unique billing code. Self-administered, outpatient drugs and cell based therapies are typically reimbursed under Medicare Part D, and cell based therapies that are administered in an inpatient hospital setting are typically reimbursed under Medicare Part A under a bundled payment. It is difficult for us to predict how Medicare coverage and reimbursement policies will be applied to our products in the future and coverage and reimbursement under different federal healthcare programs are not always consistent. Medicare reimbursement rates may also reflect budgetary constraints placed on the Medicare program.
Third party payors often rely upon Medicare coverage policies and payment limitations in setting their own reimbursement rates. These coverage policies and limitations may rely, in part, on compendia listings for approved therapeutics. Our inability to promptly obtain relevant compendia listings, coverage, and adequate reimbursement from both government-funded and private payors for new cell based therapies that we develop and for which we obtain regulatory approval could have a material adverse effect on our operating results, our ability to raise capital needed to commercialize products and our financial condition.
We expect that these and other healthcare reform measures that may be adopted in the future, may result in more rigorous coverage criteria and lower reimbursement, and in additional downward pressure on the price that we receive for any approved product. Any reduction in reimbursement from Medicare or other government-funded programs may result in a similar reduction in payments from private payors. The implementation of cost containment measures or other healthcare reforms may prevent us from being able to generate revenue, attain profitability or commercialize our cell based therapies, once marketing approval is obtained.
We believe that the efforts of governments and third party payors to contain or reduce the cost of healthcare and legislative and regulatory proposals to broaden the availability of healthcare will continue to affect the business and financial condition of pharmaceutical and biopharmaceutical companies. A number of legislative and regulatory changes in the healthcare system in the U.S. and other major healthcare markets have been proposed, and such efforts have expanded substantially in recent years. These developments could, directly or indirectly, affect our ability to sell our products, if approved, at a favorable price. For example, in the United States, in 2010, the U.S. Congress passed the ACA, a sweeping law intended to broaden access to health insurance, reduce or constrain the growth of health spending, enhance remedies against fraud and abuse, add new transparency requirements for the healthcare and health insurance industries, impose new taxes and fees on the health industry and impose additional policy reforms. Among the provisions of the ACA addressing coverage and reimbursement of pharmaceutical products, of importance to our potential therapeutic candidates are the following:
| ● | increases to pharmaceutical manufacturer rebate liability under the Medicaid Drug Rebate Program due to an increase in the minimum basic Medicaid rebate on most branded prescription drugs and the application of Medicaid rebate liability to drugs used in risk-based Medicaid managed care plans; |
| ● | the expansion of the 340B Drug Pricing Program to require discounts for “covered outpatient drugs” sold to certain children’s hospitals, critical access hospitals, freestanding cancer hospitals, rural referral centers, and sole community hospitals; |
| ● | requirements imposed on pharmaceutical companies are required to offer discounts on brand-name cell based therapy to patients who fall within the Medicare Part D coverage gap, commonly referred to as the “Donut Hole”; |
36
| ● | requirements imposed on pharmaceutical companies to pay an annual non-tax-deductible fee to the federal government based on each company’s market share of prior year total sales of branded drugs to certain federal healthcare programs, such as Medicare, Medicaid, Department of Veterans Affairs and Department of Defense; and |
| ● | for products classified as biologics, marketing approval for a follow-on biologic product may not become effective until 12 years after the date on which the reference innovator biologic product was first licensed by the FDA, with a possible six-month extension for pediatric products. After this exclusivity ends, it may be possible for biosimilar manufacturers to enter the market, which is likely to reduce the pricing for the innovator product and could affect our profitability if our products are classified as biologics. |
Separately, pursuant to the health reform legislation and related initiatives, the Centers for Medicare and Medicaid Services, or CMS, is working with various healthcare providers to develop, refine, and implement Accountable Care Organizations, or ACOs, and other innovative models of care for Medicare and Medicaid beneficiaries, including the Bundled Payments for Care Improvement Initiative, the Comprehensive Primary Care Initiative, the Duals Demonstration, and other models. The continued development and expansion of ACOs and other innovative models of care will have an uncertain impact on any future reimbursement we may receive for approved therapeutics administered by these organizations.
The healthcare industry is heavily regulated in the U.S. at the federal, state, and local levels, and our failure to comply with applicable requirements may subject us to penalties and negatively affect our financial condition.
As a healthcare company, our operations, clinical trial activities and interactions with healthcare providers may be subject to extensive regulation in the U.S., particularly if we receive FDA approval for any of its products in the future. For example, if we receive FDA approval for a product for which reimbursement is available under a federal healthcare program (e.g., Medicare, Medicaid), it would be subject to a variety of federal laws and regulations, including those that prohibit the filing of false or improper claims for payment by federal healthcare programs (e.g. the federal False Claims Act), prohibit unlawful inducements for the referral of business reimbursable by federal healthcare programs (e.g. the federal Anti-Kickback Statute), and require disclosure of certain payments or other transfers of value made to U.S.-licensed physicians and teaching hospitals or Open Payments. We are not able to predict how third parties will interpret these laws and apply applicable governmental guidance and may challenge our practices and activities under one or more of these laws. If our past or present operations are found to be in violation of any of these laws, we could be subject to civil and criminal penalties, which could hurt our business, our operations and financial condition.
The federal Anti-Kickback Statute prohibits, among other things, any person or entity, from knowingly and willfully offering, paying, soliciting or receiving any remuneration, directly or indirectly, overtly or covertly, in cash or in kind, to induce or in return for purchasing, leasing, ordering or arranging for the purchase, lease or order of any item or service reimbursable under Medicare, Medicaid or other federal healthcare programs. The term remuneration has been interpreted broadly to include anything of value. The Anti-Kickback Statute has been interpreted to apply to arrangements between pharmaceutical manufacturers on one hand and prescribers, purchasers, and formulary managers on the other. There are a number of statutory exceptions and regulatory safe harbors protecting some common activities from prosecution. The exceptions and safe harbors are drawn narrowly and practices that involve remuneration that may be alleged to be intended to induce prescribing, purchasing or recommending may be subject to scrutiny if they do not qualify for an exception or safe harbor. Failure to meet all of the requirements of a particular applicable statutory exception or regulatory safe harbor does not make the conduct per se illegal under the Anti-Kickback Statute. Instead, the legality of the arrangement will be evaluated on a case-by-case basis based on a cumulative review of all of its facts and circumstances. Our practices may not in all cases meet all of the criteria for protection under a statutory exception or regulatory safe harbor.
Additionally, the intent standard under the Anti-Kickback Statute was amended by the ACA, to a stricter standard such that a person or entity no longer needs to have actual knowledge of the statute or specific intent to violate it in order to have committed a violation. In addition, the ACA codified case law that a claim including items or services resulting from a violation of the federal Anti- Kickback Statute constitutes a false or fraudulent claim for purposes of the federal FCA.
37
The civil monetary penalties statute imposes penalties against any person or entity that, among other things, is determined to have presented or caused to be presented a claim to a federal healthcare program that the person knows or should know is for an item or service that was not provided as claimed or is false or fraudulent.
Federal false claims and false statement laws, including the federal FCA, prohibit, among other things, any person or entity from knowingly presenting, or causing to be presented, a false or fraudulent claim for payment to, or approval by, the federal healthcare programs, including Medicare and Medicaid, or knowingly making, using, or causing to be made or used a false record or statement material to a false or fraudulent claim to the federal government. A claim includes “any request or demand” for money or property presented to the U.S. government. For instance, historically, pharmaceutical and other healthcare companies have been prosecuted under these laws for allegedly providing free product to customers with the expectation that the customers would bill federal programs for the product. Other companies have been prosecuted for causing false claims to be submitted because of the companies’ marketing of the product for unapproved, off-label, and thus generally non-reimbursable, uses.
HIPAA prohibits, among other offenses, knowingly and willfully executing a scheme to defraud any health care benefit program, including private payors, or falsifying, concealing or covering up a material fact or making any materially false, fictitious or fraudulent statement in connection with the delivery of or payment for items or services under a health care benefit program. To the extent that we act as a business associate to a healthcare provider engaging in electronic transactions, we may also be subject to the privacy and security provisions of HIPAA, as amended by HITECH, which restricts the use and disclosure of patient-identifiable health information, mandates the adoption of standards relating to the privacy and security of patient-identifiable health information, and requires the reporting of certain security breaches to healthcare provider customers with respect to such information. Additionally, many states have enacted similar laws that may impose more stringent requirements on entities like ours. Failure to comply with applicable laws and regulations could result in substantial penalties and adversely affect our financial condition and results of operations.
Many states also have similar fraud and abuse statutes or regulations that apply to items and services reimbursed under Medicaid and other state programs, or, in several states, apply regardless of the payor. Additionally, to the extent that our product is sold in a foreign country, we may be subject to similar foreign laws.
Our products, once approved, may be eligible for coverage under Medicare and Medicaid, among other government healthcare programs. Accordingly, we may be subject to a number of obligations based on their participation in these programs, such as a requirement to calculate and report certain price reporting metrics to the government, such as average sales price (ASP) and best price. Penalties may apply in some cases when such metrics are not submitted accurately and timely. Further, these prices for drugs may be reduced by mandatory discounts or rebates required by government healthcare programs or private payors and by any future relaxation of laws that presently restrict imports of drugs and biological products from countries where they may be sold at lower prices than in the United States. It is difficult to predict how Medicare coverage and reimbursement policies will be applied to our products in the future and coverage and reimbursement under different federal healthcare programs are not always consistent. Medicare reimbursement rates may also reflect budgetary constraints placed on the Medicare program.
In order to distribute products commercially, we must comply with state laws that require the registration of manufacturers and wholesale distributors of drug and biological products in a state, including, in certain states, manufacturers and distributors who ship products into the state even if such manufacturers or distributors have no place of business within the state. Some states also impose requirements on manufacturers and distributors to establish the pedigree of product in the chain of distribution, including some states that require manufacturers and others to adopt new technology capable of tracking and tracing product as it moves through the distribution chain. Several states have enacted legislation requiring pharmaceutical and biotechnology companies to establish marketing compliance programs, file periodic reports with the state, make periodic public disclosures on sales, marketing, pricing, clinical trials and other activities, and/or register their sales representatives, as well as to prohibit pharmacies and other healthcare entities from providing certain physician prescribing data to pharmaceutical and biotechnology companies for use in sales and marketing, and to prohibit certain other sales and marketing practices. All of our activities are potentially subject to federal and state consumer protection and unfair competition laws.
38
If our operations are found to be in violation of any of the federal and state healthcare laws described above or any other governmental regulations that apply to us, we may be subject to penalties, including without limitation, civil, criminal and/or administrative penalties, damages, fines, disgorgement, exclusion from participation in government programs, such as Medicare and Medicaid, injunctions, private “qui tam” actions brought by individual whistleblowers in the name of the government, or refusal to allow us to enter into government contracts, contractual damages, reputational harm, administrative burdens, diminished profits and future earnings, and the curtailment or restructuring of our operations, any of which could adversely affect our ability to operate our business and our results of operations.
Our ability to obtain reimbursement or funding from the federal government may be impacted by possible reductions in federal spending.
U.S. federal government agencies currently face potentially significant spending reductions. The Budget Control Act of 2011, or the BCA, established a Joint Select Committee on Deficit Reduction, which was tasked with achieving a reduction in the federal debt level of at least $1.2 trillion. That committee did not draft a proposal by the BCA’s deadline. As a result, automatic cuts, referred to as sequestration, in various federal programs were scheduled to take place, beginning in January 2013, although the American Taxpayer Relief Act of 2012 delayed the BCA’s automatic cuts until March 1, 2013. While the Medicare program’s eligibility and scope of benefits are generally exempt from these cuts, Medicare payments to providers and Part D health plans are not exempt. The BCA did, however, provide that the Medicare cuts to providers and Part D health plans would not exceed two percent. President Obama issued the sequestration order on March 1, 2013, and cuts went into effect on April 1, 2013. Additionally, the Bipartisan Budget Act of 2015 extended sequestration for Medicare through fiscal year 2027.
The U.S. federal budget remains in flux, which could, among other things, cut Medicare payments to providers. The Medicare program is frequently mentioned as a target for spending cuts. The full impact on our business of any future cuts in Medicare or other programs is uncertain. In addition, we cannot predict any impact President Trump’s administration and the U.S. Congress may have on the federal budget. If federal spending is reduced, anticipated budgetary shortfalls may also impact the ability of relevant agencies, such as the FDA or the National Institutes of Health, to continue to function at current levels. Amounts allocated to federal grants and contracts may be reduced or eliminated. These reductions may also impact the ability of relevant agencies to timely review and approve drug research and development, manufacturing, and marketing activities, which may delay our ability to develop, market and sell any products we may develop.
Risks Related to Doing Business in China
If we become directly subject to the recent scrutiny, criticism and negative publicity involving certain U.S.-listed Chinese companies, we may have to expend significant resources to investigate and resolve the matter which could harm our business operations, stock price and reputation and could result in a loss of your investment in our stock, especially if such matter cannot be addressed and resolved quickly.
Recently, U.S. public companies that have substantially all of their operations in China, particularly companies like us which have completed so-called reverse merger transactions, have been the subject of intense scrutiny, criticism and negative publicity by investors, short sellers, financial commentators and regulatory agencies, such as the United States Securities and Exchange Commission. Much of the scrutiny, criticism and negative publicity has centered around financial and accounting irregularities and mistakes, a lack of effective internal controls over financial accounting, inadequate corporate governance policies or a lack of adherence thereto and, in many cases, allegations of fraud. As a result of the scrutiny, criticism and negative publicity, the publicly traded stock of many U.S. listed Chinese companies has sharply decreased in value and, in some cases, has become virtually worthless. Many of these companies are now subject to shareholder lawsuits, SEC enforcement actions and are conducting internal and external investigations into the allegations. It is not clear what affect this sector-wide scrutiny, criticism and negative publicity will have on our company, our business and our stock price. If we become the subject of any unfavorable allegations, whether such allegations are proven to be true or untrue, we will have to expend significant resources to investigate such allegations and/or defend our company. This situation could be costly and time consuming and distract our management from growing our company. If such allegations are not proven to be groundless, our company and business operations will be severely impacted and your investment in our stock could be rendered worthless.
39
Adverse changes in political and economic policies of the PRC government could impede the overall economic growth of China, which could reduce the demand for our products and damage our business.
Presently, we generate our revenue in China although we intend to pursue various opportunities in the United States and our headquarters is based in the United States. Accordingly, our business, financial condition, results of operations and prospects are affected significantly by economic, political and legal developments in China. The PRC economy differs from the economies of most developed countries in many respects, including:
| ● | the higher level of government involvement; |
| ● | the early stage of development of the market-oriented sector of the economy; |
| ● | the rapid growth rate; |
| ● | the higher level of control over foreign exchange; and |
| ● | the allocation of resources. |
As the PRC economy has been transitioning from a planned economy to a more market-oriented economy, the PRC government has implemented various measures to encourage economic growth and guide the allocation of resources. While these measures may benefit the overall PRC economy, they may also have a negative effect on us or the healthcare industry in general.
Although the PRC government has in recent years implemented measures emphasizing the utilization of market forces for economic reform, the PRC government continues to exercise significant control over economic growth in China through the allocation of resources, controlling payment of foreign currency-denominated obligations, setting monetary policy and imposing policies that impact particular industries or companies in different ways.
Any adverse change in the economic conditions or government policies in China could have a material adverse effect on the overall economic growth and the level of new healthcare investments and expenditures in China, which in turn could lead to a reduction in demand for our services and consequently have a material adverse effect on our business and prospects.
Uncertainties with respect to the PRC legal system could limit the legal protections available to you and us.
We conduct substantially all of our business through our operating subsidiaries in the PRC. Our operating subsidiaries are generally subject to laws and regulations applicable to foreign investments in China and, in particular, laws applicable to foreign-invested enterprises. The PRC legal system is based on written statutes, and prior court decisions may be cited for reference but have limited precedential value. Since 1979, a series of new PRC laws and regulations have significantly enhanced the protections afforded to various forms of foreign investments in China. However, since the PRC legal system continues to rapidly evolve, the interpretations of many laws, regulations and rules are not always uniform and enforcement of these laws, regulations and rules involve uncertainties, which may limit legal protections available to you and us. In addition, any litigation in China may be protracted and result in substantial costs and diversion of resources and management attention. In addition, all of our executive officers and almost all of our directors are residents of China and not of the United States, and substantially all the assets of these persons are located outside the United States. As a result, it could be difficult for investors to affect service of process in the United States or to enforce a judgment obtained in the United States against our Chinese operations and subsidiaries.
40
The PRC government exerts substantial influence over the manner in which we must conduct our business activities.
The PRC government has exercised and continues to exercise substantial control over virtually every sector of the Chinese economy through regulation and state ownership. Our ability to operate in China may be harmed by changes in its laws and regulations. We believe that our operations in China are in material compliance with all applicable legal and regulatory requirements. However, the central or local governments of the jurisdictions in which we operate may impose new, stricter regulations or interpretations of existing regulations that would require additional expenditures and efforts on our part to ensure our compliance with such regulations or interpretations.
Accordingly, government actions in the future, including any decision not to continue to support recent economic reforms and to return to a more centrally planned economy or regional or local variations in the implementation of economic policies, could have a significant effect on economic conditions in China or particular regions thereof.
We may be unable to complete a business combination transaction efficiently or on favorable terms due to complicated merger and acquisition regulations implemented on September 8, 2006.
The recent PRC Regulation on Mergers and Acquisitions of Domestic Companies by Foreign Investors also governs the approval process by which a PRC company may participate in an acquisition of its assets or its equity interests. Depending on the structure of the transaction, the new regulation will require the Chinese parties to make a series of applications and supplemental applications to the government agencies. In some instances, the application process may require the presentation of economic data concerning a transaction, including appraisals of the target business and evaluations of the acquirer, which are designed to allow the government to assess the transaction. Government approvals will have expiration dates by which a transaction must be completed and reported to the government agencies. Compliance with the new regulations is likely to be more time consuming and expensive than in the past and the government can now exert more control over the combination of two businesses. Accordingly, due to the new regulation, our ability to engage in business combination transactions is extremely complicated, time consuming and expensive, and we may not be able to negotiate a transaction that is acceptable to our stockholders or sufficiently protect their interests in a transaction.
The new regulation allows PRC government agencies to assess the economic terms of a business combination transaction. Parties to a business combination transaction may have to submit to the Ministry of Commerce, or MOFCOM, and the other government agencies an appraisal report, an evaluation report and the acquisition agreement, all of which form part of the application for approval, depending on the structure of the transaction. The regulations also prohibit a transaction at an acquisition price obviously lower than the appraised value of the Chinese business or assets and in certain transaction structures, require that consideration must be paid within defined periods, generally not in excess of a year. The regulation also limits our ability to negotiate various terms of the acquisition, including aspects of the initial consideration, contingent consideration, holdback provisions, indemnification provisions and provisions relating to the assumption and allocation of assets and liabilities. Transaction structures involving trusts, nominees and similar entities are prohibited. Therefore, such regulation may impede our ability to negotiate and complete a business combination transaction on financial terms that satisfy our investors and protect our stockholders’ economic interests.
Under the current Enterprise Income Tax, or EIT, law, we may be classified as a “resident enterprise” of China. Such classification will likely result in unfavorable tax consequences to us and our non- PRC stockholders.
We are a holding company incorporated under the laws of Delaware. We conduct substantially all of our business through our wholly-owned and majority-owned subsidiaries, and we derive all of our income from these entities. Prior to January 1, 2008, dividends derived by foreign enterprises from business operations in China were not subject to the Chinese enterprise income tax. However, such tax exemption ceased as of January 1, 2008 and thereafter with the effectiveness of the new EIT law.
Under the EIT law, if we are not deemed to be a “resident enterprise” for Chinese tax purposes, a withholding tax at the rate of 10% would be applicable to any dividends paid by our Chinese subsidiaries to us. However, if we are deemed to be a “resident enterprise” established outside of China whose “place of effective management” is located in China, we would be classified as a resident enterprise for Chinese tax purposes and thus would be subject to an enterprise income tax rate of 25% on all of our income on a worldwide basis.
41
The regulations promulgated pursuant to the EIT law define the term “place of effective management” as “establishments that carry out substantial and overall management and control over the manufacturing and business operations, personnel, accounting, properties, etc. of an enterprise.” The State Administration of Taxation issued a SAT Circular 82 on April 22, 2009, which provides that the “place of effective management” of a Chinese-controlled overseas-incorporated enterprise is located in China if the following requirements are satisfied: (i) the senior management and core management departments in charge of its daily operations function are mainly located in the PRC; (ii) its financial and human resources decisions are subject to determination or approval by persons or bodies located in the PRC; (iii) its major assets, accounting books, company seals, and minutes and files of its board and shareholders’ meetings are located or kept in the PRC; and (iv) no less than half of the enterprise’s directors or senior management with voting rights reside in the PRC. SAT Circular 82 applies only to overseas registered enterprises controlled by PRC enterprises, not to those controlled by PRC individuals. If our non-PRC incorporated entities are deemed PRC tax residents, such entities would be subject to PRC tax under the EIT law.
We have analyzed the applicability of the EIT law and related regulations, and for each of the applicable periods presented, we have not accrued for PRC tax on such basis. In addition, although under the EIT law and the related regulations dividends paid to us by our PRC subsidiaries would qualify as “tax-exempted income,” we cannot assure you that such dividends will not be subject to a 10% withholding tax, as the PRC foreign exchange control authorities, which enforce the withholding tax, have not yet issued guidance with respect to the processing of outbound remittances to entities that are treated as resident enterprises for PRC enterprise income tax purposes. As a result of such changes, our historical operating results will not be indicative of our operating results for future periods and the value of our shares of common stock may be adversely affected. We are actively monitoring the possibility of “resident enterprise” treatment and are evaluating appropriate organizational changes to avoid this treatment, to the extent possible.
We may be subject to fines and legal sanctions if we or our Chinese employees fail to comply with PRC regulations relating to employee stock options granted by overseas listed companies to PRC citizens.
On December 25, 2006, the People’s Bank of China issued the Administration Measures on Individual Foreign Exchange Control, and its Implementation Rules were issued by the State Administration of Foreign Exchange, or SAFE, on January 5, 2007. Both took effect on February 1, 2007. Under these regulations, all foreign exchange matters involved in an employee stock holding plan, stock option plan or similar plan in which PRC citizens’ participation requires approval from the SAFE or its authorized branch. On March 28, 2007, the SAFE issued the Application Procedure for Foreign Exchange Administration for Domestic Individuals Participating in Employee Stock Holding Plans or Stock Option Plans of Overseas Listed Companies, or Notice 78. Under Notice 78, PRC individuals who participate in an employee stock option holding plan or a stock option plan of an overseas listed company are required, through a PRC domestic agent or PRC subsidiary of the overseas listed company, to register with the SAFE and complete certain other procedures. If we and our Chinese employees are granted shares or stock options pursuant to our share incentive plan they would be subject to Notice 78. However, in practice, there are significant uncertainties with regard to the interpretation and implementation of Notice 78. We are committed to complying with the requirements of Notice 78. However, we cannot provide any assurance that we or our Chinese employees will be able to qualify for or obtain any registration required by Notice 78. In particular, if we and/or our Chinese employees fail to comply with the provisions of Notice 78, we and/or our Chinese employees may be subject to fines and legal sanctions imposed by the SAFE or other PRC government authorities, as a result of which our business operations and employee option plans could be materially and adversely affected.
The new M&A Rules establish more complex procedures for some acquisitions of Chinese companies by foreign investor which could make it more difficult for us to pursue growth through acquisitions in China.
The New M&A Rules that became effective on September 8, 2006 established additional procedures and requirements that could make merger and acquisition activities by foreign investors more time-consuming and complex, including requirements in some instances that the Ministry of Commerce be notified in advance of any change- of-control transaction in which a foreign investor takes control of a PRC domestic enterprise. Complying with the requirements of the M&A Rules to complete such transactions could be time-consuming, and any required approval processes, including obtaining approval from the Ministry of Commerce, may delay or inhibit our ability to complete such transactions, which could materially adversely affect our ability to grow our business through acquisitions in China.
42
Government control of currency conversion and future movements in exchange rates may adversely affect our operations and financial results.
The value of the Renminbi, or RMB, the main currency used in China, fluctuates and is affected by, among other things, changes in China’s political and economic conditions. The conversion of RMB into foreign currencies such as the U.S. dollar have generally been based on rates set by the People’s Bank of China, which are set daily based on the previous day’s interbank foreign exchange market rates and current exchange rates on the world financial markets. Foreign exchange transactions continue to be subject to significant foreign exchange controls and require the approval of the State Administration of Foreign Exchange in China. These limitations could affect our ability to obtain foreign exchange through debt or equity financing, or to obtain foreign exchange for capital expenditures.
The Chinese government controls its foreign currency reserves through restrictions on imports and conversion of RMB into foreign currency. In July 2005, the Chinese government has adjusted its exchange rate policy from “Fixed Rate” to “Floating Rate”. Between July 2005 to December 2017, the exchange rate between the RMB and the U.S. dollar appreciated from RMB1.00 to $0.1205 to RMB1.00 to $0.1513. Any significant appreciation of the RMB may adversely affect our operations and financial results.
Risks Related to Our Securities
The price of our common stock may be volatile and fluctuate substantially, which could result in substantial losses for our stockholders.
Our common stock has been listed on the Nasdaq Capital Market under the symbol “AVCO” since November 5, 2018. Our common shares were traded previously on the OTC Market Group Inc.’s Venture Market (the “OTCQB”) since February 22, 2016, under the symbol “AVCO” since October 18, 2016 and “GTHC” prior to October 18, 2016.
The price of our common stock has been, and we expect it to continue to be, volatile. The stock market in general and the market for smaller healthcare companies in particular have experienced extreme volatility that has often been unrelated to the operating performance of particular companies. As a result of this volatility, you may not be able to sell your shares of common stock at or above the price you paid for your shares of common stock. The market price for our common stock may be influenced by many factors, including:
| ● | the success of competitive products or technologies; |
| ● | developments related to our existing or any future collaborations; |
| ● | regulatory or legal developments in the United States, China and other countries; |
| ● | developments or disputes concerning patent applications, issued patents or other proprietary rights; |
| ● | the recruitment or departure of key personnel; |
| ● | actual or anticipated changes in estimates as to financial results or recommendations by securities analysts; |
| ● | variations in our financial results or those of companies that are perceived to be similar to us; |
| ● | changes in the structure of healthcare payment systems; |
| ● | market conditions in the healthcare, pharmaceutical and biotechnology sectors; |
43
| ● | general economic, industry and market conditions; and |
| ● | the other factors described in this “Risk Factors” section. |
Future sales of our common stock or securities convertible or exchangeable for our common stock may cause our stock price to decline.
If our existing stockholders sell, or indicate an intention to sell, substantial amounts of our common stock in the public market after this offering, the price of our common stock could decline. The perception in the market that these sales may occur could also cause the price of our common stock to decline.
Up to 52,568,889 shares of our common stock are subject to a contractual lock-up for periods of up to 180 days following August 13, 2018. These shares can be sold, subject to any applicable volume limitations under federal securities laws, after the earlier of the expiration of, or release from, the lock-up period.
In addition, as of December 20, 2018, 2,670,000 shares and 578,891 shares of common stock are subject to outstanding options and warrants, respectively, which will become eligible for sale in the public market to the extent permitted by the provisions of various vesting schedules, the lock-up agreements and Rule 144 under the Securities Act. If the shares we may issue from time to time upon exercise of outstanding options are sold, or if it is perceived that they will be sold, by the award recipients in the public market, the price of our common stock could decline.
You may experience dilution of your ownership interests because of the future issuance of additional shares of our common or preferred stock or other securities that are convertible into or exercisable for our common or preferred stock.
In the future, we may issue our authorized but previously unissued equity securities, resulting in the dilution of the ownership interests of our stockholders. We are authorized to issue an aggregate of 490,000,000 shares of common stock and 10,000,000 shares of “blank check” preferred stock. We may issue additional shares of our common stock or other securities that are convertible into or exercisable for our common stock in connection with hiring or retaining employees, future acquisitions, future sales of our securities for capital raising purposes, or for other business purposes. The future issuance of any such additional shares of our common stock may create downward pressure on the trading price of the common stock. We expect we will need to raise additional capital in the near future to meet our working capital needs, and there can be no assurance that we will not be required to issue additional shares, warrants or other convertible securities in the future in conjunction with these capital raising efforts, including at a price (or exercise prices) below the price you paid for your stock.
The ability of our Board of Directors to issue additional stock may prevent or make more difficult certain transactions, including a sale or merger.
Our Board of Directors is authorized to issue up to 10,000,000 shares of preferred stock with powers, rights and preferences designated by it. Shares of voting or convertible preferred stock could be issued, or rights to purchase such shares could be issued, to create voting impediments or to frustrate persons seeking to effect a takeover or otherwise gain control of us. The ability of the Board of Directors to issue such additional shares of preferred stock, with rights and preferences it deems advisable, could discourage an attempt by a party to acquire control of us by tender offer or other means. Such issuances could therefore deprive stockholders of benefits that could result from such an attempt, such as the realization of a premium over the market price for their shares in a tender offer or the temporary increase in market price that such an attempt could cause. Moreover, the issuance of such additional shares of preferred stock to persons friendly to the Board of Directors could make it more difficult to remove incumbent managers and directors from office even if such change were to be favorable to stockholders generally.
44
Our status as an emerging growth company may result in reduced disclosure obligations.
We are an “emerging growth company,” as defined in the Jumpstart Our Business Startups Act, which we refer to as the JOBS Act, and we are eligible to take advantage of certain exemptions from various reporting and financial disclosure requirements that are applicable to other public companies, that are not emerging growth companies, including, but not limited to, (1) not being required to comply with the auditor attestation requirements of Section 404 of the Sarbanes-Oxley Act of 2002, or the Sarbanes-Oxley Act, (2) reduced disclosure obligations regarding executive compensation in our periodic reports and proxy statements, and (3) exemptions from the requirements of holding a non-binding advisory vote on executive compensation and stockholder approval of any golden parachute payments not previously approved. We intend to take advantage of these exemptions. Because of the reduced disclosure and because a portion of our business is conducted in China, investors may find investing in our common stock less attractive as a result, which could have an adverse effect on our stock price.
In addition, Section 102 of the JOBS Act also provides that an emerging growth company can take advantage of the extended transition period provided in Section 7(a)(2)(B) of the Securities Act of 1933, as amended, for complying with new or revised accounting standards. As a result, an emerging growth company can delay the adoption of certain accounting standards until those standards would otherwise apply to private companies. We elected to opt out of such extended transition period and acknowledge such election is irrevocable pursuant to Section 107 of the JOBS Act.
We could remain an emerging growth company for up to five years, or until the earliest of (1) the last day of the first fiscal year in which our annual gross revenues exceed $1.07 billion, (2) the date that we become a “large accelerated filer” as defined in Rule 12b-2 under the Exchange Act, which would occur if the market value of our ordinary shares that is held by non-affiliates exceeds $700 million as of the last business day of our most recently completed second fiscal quarter and we have been publicly reporting for at least 12 months, or (3) the date on which we have issued more than $1 billion in non-convertible debt during the preceding three-year period.
We are a “smaller reporting company,” and we cannot be certain if the reduced disclosure requirements applicable to smaller reporting companies will make our common stock less attractive to investors.
We are currently a “smaller reporting company”, meaning that we are not an investment company, an asset- backed issuer, or a majority-owned subsidiary of a parent company that is not a smaller reporting company and have a non-affiliated public float of less than $75.0 million and annual revenues of less than $50.0 million during the most recently completed fiscal year. In the event that we are still considered a “smaller reporting company,” at such time as we cease being an “emerging growth company,” we will be required to provide additional disclosure in our SEC filings. However, similar to an “emerging growth companies”, “smaller reporting companies” are able to provide simplified executive compensation disclosures in their filings; are exempt from the provisions of Section 404(b) of the Sarbanes-Oxley Act requiring that independent registered public accounting firms provide an attestation report on the effectiveness of internal control over financial reporting; and have certain other decreased disclosure obligations in their SEC filings, including, among other things, only being required to provide two years of audited financial statements in annual reports. Decreased disclosures in our SEC filings due to our status as a “smaller reporting company” may make it harder for investors to analyze our results of operations and financial prospects.
If securities or industry analysts do not publish research or reports about our business, or if they issue an adverse or misleading opinion regarding our stock, our stock price and trading volume could decline.
The trading market for our common stock will be influenced by the research and reports that industry or securities analysts publish about us or our business. We do not currently have and may never obtain research coverage by securities and industry analysts. If no or few securities or industry analysts commence coverage of us, the trading price for our stock would be negatively impacted. In the event we obtain securities or industry analyst coverage, if any of the analysts who cover us issue an adverse or misleading opinion regarding us, our business model, our intellectual property or our stock performance, or if our operating results fail to meet the expectations of analysts, our stock price would likely decline. If one or more of these analysts cease coverage of us or fail to publish reports on us regularly, we could lose visibility in the financial markets, which in turn could cause our stock price or trading volume to decline.
45
Our officers, directors and principal stockholders own a significant percentage of our stock and will be able to exert significant control over matters subject to stockholder approval.
Our officers, directors and 5% stockholders and their affiliates beneficially own a significant percentage of our outstanding common stock. As a result, these stockholders have significant influence and may be able to determine all matters requiring stockholder approval. For example, these stockholders may be able to control elections of directors, amendments of our organizational documents, or approval of any merger, sale of assets, or other major corporate transactions. This concentration of ownership could delay or prevent any acquisition of our company on terms that other stockholders may desire, and may adversely affect the market price of our common stock.
Our management will have broad discretion in the use of the net proceeds from this offering and may not use them effectively.
Our management will have broad discretion in the application of the net proceeds from this offering and our stockholders will not have the opportunity as part of their investment decisions to assess whether the net proceeds are being used appropriately. You may not agree with our decisions, and our use of the proceeds may not yield any return on your investment. Because of the number and variability of factors that will determine our use of the net proceeds from this offering, their ultimate use may vary substantially from their currently intended use. Our failure to apply the net proceeds of this offering effectively could compromise our ability to pursue our growth strategy and we might not be able to yield a significant return, if any, in our investment of these net proceeds. You will not have the opportunity to influence our decisions on how to use our net proceeds from this offering.
We may be exposed to additional risks as a result of “going public” by means of a reverse acquisition transaction.
We may be exposed to additional risks because we became a public company through a “reverse merger” transaction. There has been increased focus by government agencies on reverse merger transactions in recent years, and we may be subject to increased scrutiny by the SEC and other government agencies and holders of our securities as a result of the completion of our reverse merger transaction. Additionally, our “going public” by means of a reverse merger transaction may make it more difficult for us to obtain coverage from securities analysts of major brokerage firms following the reverse merger transaction because there may be little incentive to those brokerage firms to recommend the purchase of our common stock. Further, investment banks may be less likely to agree to underwrite secondary offerings on our behalf than they might if we became a public reporting company by means of an initial public offering because they may be less familiar with our company as a result of more limited coverage by analysts and the media, and because we became public at an early stage in our development. The failure to receive research coverage or support in the market for our shares will have an adverse effect on our ability to develop a liquid market for our common stock. The occurrence of any such event could cause our business or stock price to suffer.
We do not anticipate paying dividends on our common stock, and investors may lose the entire amount of their investment.
We have never declared or paid cash dividends on our common stock, and we do not anticipate such a declaration or payment for the foreseeable future.
We expect to use future earnings, if any, to fund business growth. Therefore, stockholders will not receive any funds absent a sale of their shares of common stock. We cannot assure stockholders of a positive return on their investment when they sell their shares, nor can we assure that stockholders will not lose the entire amount of their investment.
Applicable regulatory requirements, including those contained in and issued under the Sarbanes-Oxley Act of 2002, may make it difficult for us to retain or attract qualified officers and directors, which could adversely affect the management of our business and our ability to obtain or retain listing of our common stock on a national securities exchange.
We may be unable to attract and retain those qualified officers, directors and members of board committees required to provide for effective management because of the rules and regulations that govern publicly held companies, including, but not limited to, certifications by principal executive officers. The enactment of the Sarbanes-Oxley Act has resulted in the issuance of a series of related rules and regulations and the strengthening of existing rules and regulations by the SEC, as well as the adoption of new and more stringent rules by national securities exchanges. The perceived increased personal risk associated with these changes may deter qualified individuals from accepting roles as directors and executive officers.
46
Further, some of these changes heighten the requirements for board or committee membership, particularly with respect to an individual’s independence from the corporation and level of experience in finance and accounting matters. We may have difficulty attracting and retaining directors with the requisite qualifications. If we are unable to attract and retain qualified officers and directors, the management of our business and our ability to obtain or retain listing of our shares of common stock on any national securities exchange could be adversely affected.
Any failure to maintain effective internal control over our financial reporting could materially adversely affect us.
Section 404 of the Sarbanes-Oxley Act of 2002 requires us to include in our annual reports on Form 10-K an assessment by management of the effectiveness of our internal control over financial reporting. In addition, at such time, if any, as we are an “accelerated filer” or a “large accelerated filer,” and no longer an “emerging growth company,” our independent registered public accounting firm will have to attest to and report on management’s assessment of the effectiveness of such internal control over financial reporting. Our management assessed our internal control over financial reporting as of December 31, 2017. Based on such assessment, we concluded that our internal control over financial reporting was not effective as of December 31, 2017 to provide reasonable assurance regarding the reliability of financial reporting and the preparation of financial statements for external reporting purposes in accordance with U.S. generally accepted accounting principles. The material weaknesses we have identified are as follows:
| ● | We have not established adequate financial reporting monitoring activities to mitigate the risk of management override, specifically because there are few employees and only two officers with management functions and therefore there is lack of segregation of duties. |
| ● | There is a strong reliance on outside consultants to review and adjust the annual and quarterly financial statements, to monitor new accounting principles, and to ensure compliance with GAAP and SEC disclosure requirements. |
| ● | There is a strong reliance on the external attorneys to review and edit the annual and quarterly filings and to ensure compliance with SEC disclosure requirements. |
| ● | A formal audit committee has not been formed as of December 31, 2017. |
Our internal control over financial reporting will not prevent or detect all error and all fraud. A control system, no matter how well designed and operated, can provide only reasonable, not absolute, assurance that the control system’s objectives will be met. Because of the inherent limitations in all control systems, no evaluation of controls can provide absolute assurance that misstatements due to error or fraud will not occur or that all control issues and instances of fraud will be detected. If we are not able to comply with the requirements of Section 404 in a timely manner, if we do not remedy the current material weaknesses or if we identify additional material weaknesses in our internal controls, investors could lose confidence in the reliability of our financial statements, the market price of our stock could decline and we could be subject to sanctions or investigations by the SEC, or other regulatory authorities.
If we cannot satisfy, or continue to satisfy, the initial listing requirements and other rules of the Nasdaq Capital Market, our securities may be delisted, which could negatively impact the price of our securities and your ability to sell them.
Our common stock has been listed on the Nasdaq Capital Market under the symbol “AVCO” since November 5, 2018. In order to maintain our listing on the Nasdaq Capital Market, we are required to comply with certain rules of the applicable trading market, including those regarding minimum stockholders’ equity, minimum share price and certain corporate governance requirements. We may not be able to continue to satisfy the listing requirements and other applicable rules of the Nasdaq Capital Market. If we are unable to satisfy the criteria for maintaining our listing, our securities could be subject to delisting.
47
If our common stock is delisted from trading by the applicable trading market we could face significant consequences, including.
| ● | a limited availability for market quotations for our securities; |
| ● | reduced liquidity with respect to our securities; |
| ● | a determination that our common stock is a “penny stock,” which will require brokers trading in our common stock to adhere to more stringent rules and possibly result in a reduced level of trading activity in the secondary trading market for our common stock; |
| ● | limited amount of news and analyst coverage; and |
| ● | a decreased ability to issue additional securities or obtain additional financing in the future. |
We could be subject to securities class action litigation.
In the past, securities class action litigation has often been brought against a company following a decline in the market price of its securities. This risk is especially relevant for us because companies in our industry have experienced significant stock price volatility in recent years. If we face such litigation, it could result in substantial costs and a diversion of management’s attention and resources, which could harm our business.
Our common stock is subject to the “penny stock” rules of the SEC and the trading market in our securities is limited, which makes transactions in our stock cumbersome and may reduce the value of an investment in our stock.
The SEC has adopted Rule 3a51-1 which establishes the definition of a “penny stock,” for the purposes relevant to us, as any equity security that has a market price of less than $5.00 per share or with an exercise price of less than $5.00 per share, subject to certain exceptions. For any transaction involving a penny stock, unless exempt, Rule 15g-9 requires:
| ● | that a broker or dealer approve a person’s account for transactions in penny stocks; and |
| ● | the broker or dealer receives from the investor a written agreement to the transaction, setting forth the identity and quantity of the penny stock to be purchased. |
In order to approve a person’s account for transactions in penny stocks, the broker or dealer must:
| ● | obtain financial information and investment experience objectives of the person; and |
| ● | make a reasonable determination that the transactions in penny stocks are suitable for that person and the person has sufficient knowledge and experience in financial matters to be capable of evaluating the risks of transactions in penny stocks. |
The broker or dealer must also deliver, prior to any transaction in a penny stock, a disclosure schedule prescribed by the SEC relating to the penny stock market, which, in highlight form:
| ● | sets forth the basis on which the broker or dealer made the suitability determination; and |
| ● | that the broker or dealer received a signed, written agreement from the investor prior to the transaction. |
48
Disclosure also has to be made about the risks of investing in penny stocks in both public offerings and in secondary trading and about the commissions payable to both the broker-dealer and the registered representative, current quotations for the securities and the rights and remedies available to an investor in cases of fraud in penny stock transactions. Finally, monthly statements have to be sent disclosing recent price information for the penny stock held in the account and information on the limited market in penny stocks.
Generally, brokers may be less willing to execute transactions in securities subject to the “penny stock” rules. This may make it more difficult for investors to dispose of our common stock and cause a decline in the market value of our stock.
CAUTIONARY NOTE REGARDING FORWARD-LOOKING STATEMENTS
This prospectus contains forward-looking statements, including, without limitation, in the sections captioned “Risk Factors”, “Management’s Discussion and Analysis of Financial Condition and Results of Operations”, and “Business”. Any and all statements contained in this prospectus that are not statements of historical fact may be deemed forward-looking statements. Terms such as “may,” “might,” “would,” “should,” “could,” “project,” “estimate,” “pro-forma,” “predict,” “potential,” “strategy,” “anticipate,” “attempt,” “develop,” “plan,” “help,” “believe,” “continue,” “intend,” “expect,” “future,” and terms of similar import (including the negative of any of the foregoing) may be intended to identify forward-looking statements. However, not all forward-looking statements may contain one or more of these identifying terms. Forward-looking statements in this prospectus may include, without limitation, statements regarding (i) the plans and objectives of management for future operations, (ii) a projection of income (including income/loss), earnings (including earnings/loss) per share, capital expenditures, dividends, capital structure or other financial items, (iii) our future financial performance, including any such statement contained in a discussion and analysis of financial condition by management or in the results of operations included pursuant to the rules and regulations of the SEC, and (iv) the assumptions underlying or relating to any statement described in points (i), (ii) or (iii) above.
The forward-looking statements are not meant to predict or guarantee actual results, performance, events or circumstances and may not be realized because they are based upon our current projections, plans, objectives, beliefs, expectations, estimates and assumptions and are subject to a number of risks and uncertainties and other influences, many of which we have no control over. Actual results and the timing of certain events and circumstances may differ materially from those described by the forward-looking statements as a result of these risks and uncertainties. Factors that may influence or contribute to the inaccuracy of the forward-looking statements or cause actual results to differ materially from expected or desired results may include, without limitation:
| ● | Our ability to attract and retain management; |
| ● | Our ability to raise capital when needed and on acceptable terms and conditions; |
| ● | The intensity of competition; |
| ● | General economic conditions; |
| ● | Changes in regulations; |
| ● | Whether the market for healthcare services continues to grow, and, if it does, the pace at which it may grow; and |
| ● | Our ability to compete against large competitors in a rapidly changing market. |
Readers are cautioned not to place undue reliance on forward-looking statements because of the risks and uncertainties related to them and to the risk factors. We disclaim any obligation to update the forward-looking statements contained in this prospectus to reflect any new information or future events or circumstances or otherwise, except as required by law.
Readers should read this prospectus in conjunction with the discussion under the caption “Risk Factors” in this prospectus and our consolidated financial statements and the related notes incorporated by reference.
49
Unless we otherwise indicate in a prospectus supplement, we currently intend to use the net proceeds from the sale of our securities for acquisitions and working capital purposes.
More detailed information regarding the use of proceeds from the sale of securities, including any determinable milestones at the applicable time, will be described in any applicable prospectus supplement. We may also, from time to time, issue securities otherwise than pursuant to a prospectus supplement to this prospectus.
We have never declared or paid any cash dividends on our common stock. We currently intend to retain all of our future earnings, if any, to finance the growth and development of our business. We do not intend to pay cash dividends to holders of our common stock in the foreseeable future.
We may offer and issue from time to time common stock, preferred stock, warrants to purchase common stock or preferred stock and units, or any combination thereof, up to an aggregate initial offering price of up to $50,000,000 in one or more transactions under this shelf prospectus. The price of securities offered will depend on a number of factors that may be relevant at the time of offer. See “Plan of Distribution.”
Our common stock has been listed on the Nasdaq Capital Market under the symbol “AVCO” since November 5, 2018. Our common shares were traded previously on the OTC Market Group Inc.’s Venture Market (the “OTCQB”) since February 22, 2016, under the symbol “AVCO” since October 18, 2016 and “GTHC” prior to October 18, 2016.
The following tables sets forth, for the periods indicated, the high and low trading prices of the common stock as reported on the Nasdaq Capital Market and OTCQB prior to the filing of this prospectus.
| OTCQB (U.S. Dollars) | NASDAQ (U.S. Dollars) | |||||||||||||||
| Period | High | Low | High | Low | ||||||||||||
| 2016 | ||||||||||||||||
| First Quarter (from February 22, 2016 to March 31, 2016) | 0.16 | 0.16 | ||||||||||||||
| Second Quarter | 0.16 | 0.04 | ||||||||||||||
| Third Quarter | 0.04 | 0.04 | ||||||||||||||
| Fourth Quarter | 3.00 | 0.04 | ||||||||||||||
| 2017 | ||||||||||||||||
| First Quarter | 5.00 | 1.00 | ||||||||||||||
| Second Quarter | 1.49 | 0.51 | ||||||||||||||
| Third Quarter | 3.50 | 0.51 | ||||||||||||||
| Fourth Quarter | 4.60 | 1.35 | ||||||||||||||
| 2018 | ||||||||||||||||
| First Quarter | 3.97 | 0.98 | ||||||||||||||
| Second Quarter | 3.30 | 1.45 | ||||||||||||||
| Third Quarter | 2.90 | 2.11 | ||||||||||||||
| Fourth Quarter (from October 1, 2018 to November 4, 2018) | 2.80 | 2.40 | ||||||||||||||
| Fourth Quarter (from November 5, 2018 to December 31, 2018) | 3.15 | 2.05 | ||||||||||||||
| 2019 | ||||||||||||||||
| First Quarter (from January 1, 2019 to January 11, 2019) | 3.40 | 2.13 | ||||||||||||||
50
We have authorized capital stock consisting of 490,000,000 shares of common stock, par value $0.0001 per share, and 10,000,000 shares of preferred stock, par value $0.0001 per share. As of December 20, 2018, we had 74,310,751 shares of common stock issued and outstanding, and no shares of preferred stock issued and outstanding.
Common Stock
All outstanding shares of common stock are of the same class and have equal rights and attributes. The holders of common stock are entitled to one vote per share on all matters submitted to a vote of stockholders of the company. All stockholders are entitled to share equally in dividends, if any, as may be declared from time to time by the Board of Directors out of funds legally available. In the event of liquidation, the holders of common stock are entitled to share ratably in all assets remaining after payment of all liabilities. The stockholders do not have cumulative or preemptive rights.
Preferred Stock
Our Certificate of Incorporation authorizes the issuance of up to 10,000,000 shares of preferred stock with designations, rights and preferences determined from time to time by our Board of Directors. Accordingly, our Board of Directors is empowered, without stockholder approval, to issue preferred stock with dividend, liquidation, conversion, voting, or other rights which could adversely affect the voting power or other rights of the holders of the common stock. In the event of issuance, the preferred stock could be utilized, under certain circumstances, as a method of discouraging, delaying or preventing a change in control of our company, which is sometimes referred to in corporate parlance as a “poison pill”.
Options and Restricted Stock
As of December 20, 2018, options to purchase 2,670,000 shares of our common stock were outstanding.
Warrants
As of December 20, 2018, warrants to purchase 578,891 shares of our common stock were outstanding.
Other Convertible Securities
As of December 20, 2018, other than the securities described above, we do not have any outstanding convertible securities.
Stockholder Action by Written Consent
Any action required or permitted to be taken at any annual or special meetings of the stockholders of the company may be taken without a meeting, without prior notice and without a vote, by a consent or consents in writing, setting forth the action so taken, (a) signed by stockholders of the company holding not less than the minimum number of votes that would be necessary to authorize or take such action at a meeting at which all the shares of the company entitled to vote thereon were present and voted and (b) delivered to the company in accordance with Section 228 of the DGCL.
Anti-Takeover Effects of Provisions of our Certificate of Incorporation, our Bylaws and Delaware Law
Some provisions of Delaware law, our certificate of incorporation and our bylaws contain provisions that could make the following transactions more difficult: acquisition of us by means of a tender offer; acquisition of us by means of a proxy contest or otherwise; or removal of our incumbent officers and directors. It is possible that these provisions could make it more difficult to accomplish or could deter transactions that stockholders may otherwise consider to be in their best interest or in our best interests, including transactions that might result in a premium over the price of our common stock.
51
These provisions, summarized below, are expected to discourage coercive takeover practices and inadequate takeover bids. These provisions are also designed to encourage persons seeking to acquire control of us to first negotiate with our board of directors. We believe that the benefits of increased protection of our potential ability to negotiate with the proponent of an unfriendly or unsolicited proposal to acquire or restructure us outweigh the disadvantages of discouraging these proposals because negotiation of these proposals could result in an improvement of their terms.
Delaware Anti-Takeover Statute
We are subject to Section 203 of the Delaware General Corporation Law, which regulates corporate takeovers. The existence of this provision may have an anti-takeover effect with respect to transactions not approved in advance by the board of directors, such as discouraging takeover attempts that might result in a premium over the price of our common stock.
Undesignated Preferred Stock
The ability to authorize undesignated preferred stock makes it possible for our board of directors to issue preferred stock with voting or other rights or preferences that could impede the success of any attempt to change control. These and other provisions may have the effect of deterring hostile takeovers or delaying changes in control or management.
Transfer Agent
The stock transfer agent for our securities is Vstock Transfer, LLC, 18 Lafayette Place, Woodmere, NY 11598, (212) 828-8436.
We may issue warrants for the purchase of shares of our common stock or preferred stock. We may issue warrants independently or together with other securities, and the warrants may be attached to or separate from any offered securities. Each series of warrants will be issued under a separate warrant agreement to be entered into between us and the investors or a warrant agent. The following summary of material provisions of the warrants and warrant agreements are subject to, and qualified in their entirety by reference to, all the provisions of the warrant agreement and warrant certificate applicable to a particular series of warrants. The terms of any warrants offered under a prospectus supplement may differ from the terms described below. We urge you to read the applicable prospectus supplement and any related free writing prospectus, as well as the complete warrant agreements and warrant certificates that contain the terms of the warrants.
The particular terms of any issue of warrants will be described in the prospectus supplement relating to the issue. Those terms may include:
| ● | the number of shares of common stock or preferred stock purchasable upon the exercise of warrants to purchase such shares and the price at which such number of shares may be purchased upon such exercise; |
| ● | the designation, stated value and terms (including, without limitation, liquidation, dividend, conversion and voting rights) of the series of preferred stock purchasable upon exercise of warrants to purchase preferred stock; |
| ● | the date, if any, on and after which the warrants, preferred stock or common stock will be separately transferable; |
| ● | the terms of any rights to redeem or call the warrants; |
| ● | the date on which the right to exercise the warrants will commence and the date on which the right will expire; |
| ● | United States federal income tax consequences applicable to the warrants; and |
| ● | any additional terms of the warrants, including terms, procedures, and limitations relating to the exchange, exercise and settlement of the warrants. |
52
Holders of equity warrants will not be entitled to:
| ● | vote, consent or receive dividends; |
| ● | receive notice as stockholders with respect to any meeting of stockholders for the election of our directors or any other matter; or |
| ● | exercise any rights as stockholders of Avalon Globocare. |
Each warrant will entitle its holder to purchase the principal amount of the number of shares of preferred stock or common stock at the exercise price set forth in, or calculable as set forth in, the applicable prospectus supplement. Unless we otherwise specify in the applicable prospectus supplement, holders of the warrants may exercise the warrants at any time up to the specified time on the expiration date that we set forth in the applicable prospectus supplement. After the close of business on the expiration date, unexercised warrants will become void.
A holder of warrant certificates may exchange them for new warrant certificates of different denominations, present them for registration of transfer and exercise them at the corporate trust office of the warrant agent or any other office indicated in the applicable prospectus supplement. Until any warrants to purchase common stock or preferred stock are exercised, the holders of the warrants will not have any rights of holders of the underlying common stock or preferred stock, including any rights to receive dividends or payments upon any liquidation, dissolution or winding up on the common stock or preferred stock, if any.
We may issue units consisting of any combination of the other types of securities offered under this prospectus in one or more series. We may evidence each series of units by unit certificates that we will issue under a separate agreement. We may enter into unit agreements with a unit agent. Each unit agent will be a bank or trust company that we select. We will indicate the name and address of the unit agent in the applicable prospectus supplement relating to a particular series of units.
The following description, together with the additional information included in any applicable prospectus supplement, summarizes the general features of the units that we may offer under this prospectus. You should read any prospectus supplement and any free writing prospectus that we may authorize to be provided to you related to the series of units being offered, as well as the complete unit agreements that contain the terms of the units. Specific unit agreements will contain additional important terms and provisions and we will file as an exhibit to the registration statement of which this prospectus is a part, or will incorporate by reference from another report that we file with the SEC, the form of each unit agreement relating to units offered under this prospectus.
If we offer any units, certain terms of that series of units will be described in the applicable prospectus supplement, including, without limitation, the following, as applicable:
| ● | the title of the series of units; |
| ● | identification and description of the separate constituent securities comprising the units; |
| ● | the price or prices at which the units will be issued; |
| ● | the date, if any, on and after which the constituent securities comprising the units will be separately transferable; |
| ● | a discussion of certain United States federal income tax considerations applicable to the units; and |
| ● | any other terms of the units and their constituent securities. |
The applicable prospectus supplement may describe certain U.S. federal income tax consequences to an investor who is a U.S. person (within the meaning of the U.S. Internal Revenue Code).
53
We may sell the securities in one or more of the following ways (or in any combination) from time to time:
| ● | through underwriters or dealers; |
| ● | directly to a limited number of purchasers or to a single purchaser; |
| ● | through agents; |
| ● | through a combination of any such methods; or |
| ● | through any other methods described in a prospectus supplement. |
The prospectus supplement will state the terms of the offering of the securities, including:
| ● | the name or names of any underwriters, dealers or agents; |
| ● | the purchase price of such securities and the proceeds to be received by us, if any; |
| ● | any underwriting discounts or agency fees and other items constituting underwriters’ or agents’ compensation; |
| ● | any initial public offering price; |
| ● | any discounts or concessions allowed or reallowed or paid to dealers; and |
| ● | any securities exchanges on which the securities may be listed. |
Any initial public offering price and any discounts or concessions allowed or reallowed or paid to dealers may be changed from time to time.
If we use underwriters in the sale, the securities will be acquired by the underwriters for their own account and may be resold from time to time in one or more transactions, including:
| ● | negotiated transactions; |
| ● | at a fixed public offering price or prices, which may be changed; |
| ● | at market prices prevailing at the time of sale; |
| ● | at prices related to prevailing market prices; or |
| ● | at negotiated prices. |
Unless otherwise stated in a prospectus supplement, the obligations of the underwriters to purchase any securities will be conditioned on customary closing conditions and the underwriters will be obligated to purchase all of such series of securities, if any are purchased.
We may sell the securities through agents from time to time. The prospectus supplement will name any agent involved in the offer or sale of the securities and any commissions we pay to them. Generally, any agent will be acting on a best efforts basis for the period of its appointment.
We may authorize underwriters, dealers or agents to solicit offers by certain purchasers to purchase the securities from us at the public offering price set forth in the prospectus supplement pursuant to delayed delivery contracts providing for payment and delivery on a specified date in the future. The contracts will be subject only to those conditions set forth in the prospectus supplement, and the prospectus supplement will set forth any commissions we pay for solicitation of these contracts.
Underwriters and agents may be entitled under agreements entered into with us to indemnification by us against certain civil liabilities, including liabilities under the Securities Act or to contribution with respect to payments which the underwriters or agents may be required to make. Underwriters and agents may be customers of, engage in transactions with, or perform services for us and our affiliates in the ordinary course of business.
Each series of securities will be a new issue of securities and will have no established trading market other than our common stock, which is listed on the Nasdaq Capital Market. Any underwriters to whom securities are sold for public offering and sale may make a market in the securities, but such underwriters will not be obligated to do so and may discontinue any market making at any time without notice. The securities, other than the common stock, may or may not be listed on a national securities exchange.
54
WHERE YOU CAN FIND MORE INFORMATION
We have filed with the SEC a registration statement on Form S-3 under the Securities Act with respect to the securities described in this prospectus and any accompanying prospectus supplement, as applicable. This prospectus and any accompanying prospectus supplement, which constitute a part of that registration statement, do not contain all of the information set forth in that registration statement and its exhibits. For further information with respect to us and our securities, you should consult the registration statement and its exhibits.
We are subject to the information and periodic reporting requirements of the Exchange Act and, accordingly, we file annual reports containing financial statements audited by an independent registered public accounting firm, quarterly reports containing unaudited financial data, current reports and other reports and information with the SEC. You may read and copy all or any portion of the registration statement without charge at the public reference room of the SEC at 100 F Street, N. E., Washington, D.C. 20549. Copies of the registration statement may be obtained from the SEC at prescribed rates from the public reference room of the SEC at such address. You may obtain information regarding the operation of the public reference room by calling 1-800-SEC-0330. In addition, registration statements and certain other filings made with the SEC electronically are publicly available through the SEC’s web site at http://www.sec.gov. The registration statement, including all exhibits and amendments thereto, has been filed electronically with the SEC.
The SEC allows us to “incorporate by reference” into this prospectus the documents we file with, or furnish to, it, which means that we can disclose important information to you by referring you to these documents. The information that we incorporate by reference into this prospectus forms a part of this prospectus, and information that we file later with the SEC automatically updates and supersedes any information in this prospectus. We incorporate by reference into this prospectus the documents listed below:
| ● | Annual Report on Form 10-K for the fiscal year ended December 31, 2017, including any amendments, initially filed with the SEC on March 13, 2018; |
| ● | Preliminary Information Statement of Schedule 14C filed with the SEC on March 16, 2018; |
| ● | Definitive Information Statement of Schedule 14C filed with the SEC on April 3, 2018; |
| ● | Current Report on Form 8-K, filed with the SEC on April 4, 2018, with respect to compensatory arrangements of certain officers; |
| ● | Current Report on Form 8-K, filed with the SEC on April 18, 2018, with respect to entry into a material definitive agreement and unregistered sales of equity securities, and the Amendment No. 1, Amendment No. 2, Amendment No. 3 to this Current Report on Form 8-K/A filed with on April 26, May 1, and May 31, 2018, respectively; |
| ● | Quarterly Report on Form 10-Q, including any amendments, initially filed with the SEC on May 11, 2018; |
| ● | Current Report on Form 8-K, filed with the SEC on May 14, 2018, with respect to certain press release; |
55
| ● | Current Report on Form 8-K, filed with the SEC on June 6, 2018, with respect to entry into a material definitive agreement, unregistered sales of equity securities, and appointment of new director; |
| ● | Current Report on Form 8-K, filed with the SEC on July 10, 2018, with respect to entry into a material definitive agreement, unregistered sales of equity securities, departure of officer and appointment of new director; |
| ● | Current Report on Form 8-K, filed with the SEC on July 31, 2018, with respect to entry into a material definitive agreement, unregistered sales of equity securities, departure of director and appointment of new director; |
| ● | Quarterly Report on Form 10-Q, including any amendments, initially filed with the SEC on August 14, 2018; |
| ● | Current Report on Form 8-K, filed with the SEC on August 15, 2018, with respect to certain press release. |
| ● | Current Report on Form 8-K, filed with the SEC on October 29, 2018, with respect to entry into a material definitive agreement; |
| ● | the description of our common stock contained in our Registration Statement on Form 8-A filed with the SEC on November 2, 2018, including any amendments or reports filed for the purpose of updating that description (File No. 001-38728); |
| ● | Quarterly Report on Form 10-Q, including any amendments, initially filed with the SEC on November 13, 2018; and |
| ● | Current Report on Form 8-K, filed with the SEC on November 14, 2018, with respect to certain press release. | |
| ● | Current Report on Form 8-K, filed with the SEC on January 3, 2019, with respect to Departure of Directors or Certain Officers; Election of Directors; Appointment of Certain Officers; Compensatory Arrangements of Certain Officers. |
All documents filed by us pursuant to Sections 13(a), 13(c), 14 or 15(d) of the Exchange Act subsequent to the date of this prospectus and prior to the termination of the offering of the securities offered by this prospectus are incorporated by reference into this prospectus and form part of this prospectus from the date of filing or furnishing of these documents. Notwithstanding the foregoing, unless specifically statements to the contrary, none of the information that is not deemed “filed” with the SEC, including information furnished under Item 2.02 or 7.01 of the Current Report on Form 8-K, will be incorporated by reference into, or otherwise included in, this prospectus.
Any statement contained in a document that is incorporated by reference into this prospectus will be deemed to be modified or superseded for the purposes of this prospectus to the extent that a statement contained in this prospectus, or in any other subsequently filed document which also is or is deemed to be incorporated by reference into this prospectus, modifies or supersedes that statement. The modifying or superseding statement does not need to state that it has modified or superseded a prior statement or include any other information set forth in the document that it modifies or supersedes.
Upon request, we will provide, without charge, to each person who receives this prospectus, a copy of any or all of the documents incorporated by reference (other than exhibits to the documents that are not specifically incorporated by reference in the documents). Please direct written or oral requests for copies to our Corporate Secretary at 4400 Route 9 South, Suite 3100, Freehold, New Jersey 07728 or by calling 732-780-4400.
There has been no material changes which have occurred since the end of the latest fiscal year for which certified financial statements were included in the latest annual report on from 10-K to security holders and which have not been described in a report on Form 10-Q or Form 8-K filed under the Exchange Act.
56
Ortoli Rosenstadt LLP is acting as counsel to our company regarding U.S. securities law matters. The current address of Ortoli Rosenstadt LLP is 366 Madison Avenue, 3rd Floor, New York, NY 10017.
RBSM LLP, an independent registered public accounting firm, has audited our consolidated financial statements included in our Annual Report on Form 10-K for the year ended December 31, 2017, as set forth in their report dated March 12, 2018 (which contains an explanatory paragraph about our ability to continue as a going concern), which is incorporated by reference in this prospectus and elsewhere in the registration statement. Our consolidated financial statements are incorporated by reference in reliance on RBSM LLP’s report, given on their authority as experts in accounting and auditing.
57
Up to $20,000,000
Common Stock
PROSPECTUS SUPPLEMENT
Jefferies
December 13, 2019